
11q24-q25 缺失2 例临床表型及文献复习
2012-12-23 04:22陈龙霞詹国栋王来栓程国强黄国英周文浩
中国循证儿科杂志 2012年4期
杨 琳 陈龙霞 詹国栋 王来栓 程国强 马 端 黄国英 周文浩
1 病例资料
例1 男,1 d,因发现“喂养困难,胎便异常1 h”入院。母亲32 岁,G1P1,孕37+5周,剖宫产,出生后1 和5 min Apgar 评分均为9 分,出生体重2 795 g(P73)。母亲产前B超检查示脐带绕颈2 周。查体:头围43 cm,身长50 cm,低位耳,四肢短小,双上肢肌张力稍高,双侧反射亢进,双侧Babinski 征(-)。心肺查体未见异常。超声心动图示卵圆孔未闭,动脉导管未闭。胸部X 线示心影过大。头颅MRI示右侧尾状核头部稍高信号(图1A)。头颅B 超示右侧尾状核头部、右侧脉络膜体部囊性占位;腹部B 超示右侧睾丸位于腹股沟内环处。脑电图示不正常脑电图,两侧脑半球背景电活动偏慢,2 ~3 Hz 的δ 波为主和部分4 ~5 Hz 的θ 波,且电压较平坦。脑电地形图示脑电功率为普遍性低功率改变。听力筛查未通过。血、尿和粪常规检查未见异常,肝、肾功能检查未见异常。无惊厥发作。父母体健,否认家族中糖尿病、先天性心脏病史,否认近亲结婚。母亲否认孕期感染史、致畸剂接触史和传染病接触史。
例2 女,2 d,因“纳差2 d”入院。母亲27 岁,G1P1,孕38+5周,剖宫产,出生后1 min Apgar 评分为10 分,出生体重3 430 g(P85)。产前B 超检查示羊水过多。查体:头围35 cm,身长50 cm,小鼻,短人中,舌喜外伸,胸前见一赘生物,脐疝,心前区杂音,脾大。超声心动图示动脉导管未闭、卵圆孔未闭(图1B)。头颅B 超示双侧脑室扩大、不对称,左侧脉络膜头部囊性占位。头颅MRI 示脑发育不全(图1C)。腹部B 超示脾脏肿大,膀胱未见充盈。听力筛查未通过。脑干诱发电位示双耳重度听力损害。血常规,肝、肾功能,凝血功能检查未见异常。无惊厥发作。母亲否认孕期感染史、致畸剂接触史,否认近亲结婚及相关遗传病家族史。
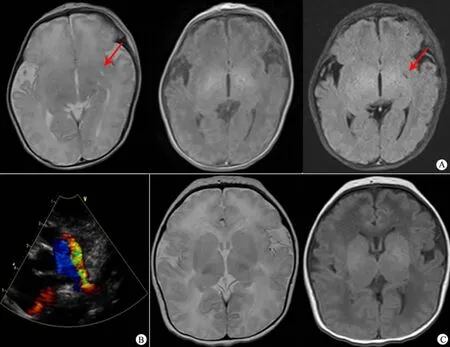
图1 2 例患儿MRI 和超声心动图所见Fig 1 MRI and echocardiography findings in 2 cases
考虑2 例患儿均存在多系统先天性畸形,在征得患儿家属知情同意后行SNP 芯片检测。取外周静脉血2 mL,经EDTA 抗凝,使用FUJI 公司QuickGene-Mini80 及DNA 全血试剂盒,提取基因组DNA。检测DNA 浓度及A 值,电泳检测DNA 降解程度,取3 ~5 μg DNA 进行芯片检测。采用cytogenetic whole genome 2.7 M 芯片(Affymetrix 公司)进行检测,采用Chromosome Analysis Suite (ChAS)软件进行数据结果的分析。
具有潜在临床意义的CNVs 片段的筛选步骤:①ChAs软件分析的结果中,选择重复片段>150 kb,缺失片段>50 kb 的CNVs 数据行下一步分析;②对照原始图像除外假阳性结果;③除外在国际基因组拷贝数变异多态性数据库(Database of Genomic Variation,DGV)报道的正常人群的CNVs(片段至少有80%以上重合,且重复或缺失类型相同);④除外片段区域内不包含基因的CNVs;⑤选取常染色体上>2.5 Mb 的CNVs。
表1 为例1、2 CNVs 起始位点及大小。图2 显示,例1和2 为11 号染色体长臂的缺失,缺失大小分别为7.5(图2A)和5.6 Mb(图2C);例1 还存在12 号染色体短臂较大片段重复11.5 Mb(图2B),例2 具有11 号染色体短臂的大片段重复32.5 Mb(图2C)。
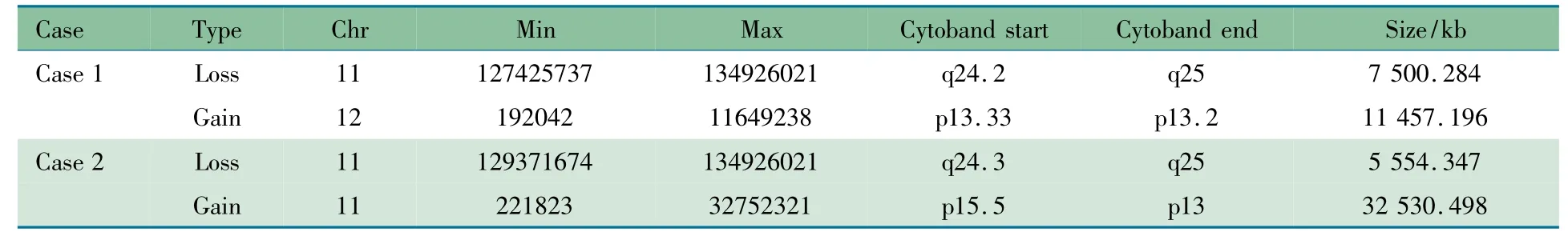
表1 2 例患儿检测到CNVs 的基本特征Tab 1 Characteristics of rare CNVs with potential clinical significance of 2 cases

图2 2 例患儿的缺失和重复Fig 2 Deletions and duplications of 2 patients by affymetrix array
2 讨论
本文2 例为11q24-q25 缺失,均在既往文献Jacobsen综合征(JBS)关键区域和缺失大小范围之内,均为末端缺失;例2 在11 号染色体短臂的大片段重复包含了Beckwith-Wiedemann 综合征(BWS)的关键区段11p15.5,在既往报道的JBS 中均未出现如例1 检测到的12 号染色体短臂的较大片段重复。
JBS 是一种罕见的由11q 远端缺失导致的以精神发育迟缓并多发畸形为主要表现的综合征[1]。1973 年首次报道[2]以来,迄今已报道260 余例。国外资料显示新生儿JBS 的发病率约为1/100 000,男∶女约为1∶2[1],国内未见相关报道。综合目前JBS 的文献,基因缺失的大小为5 ~20 Mb,关键区域为11q23-11qter,以末端缺失为主,很少一部分为中间缺失[3~6]。
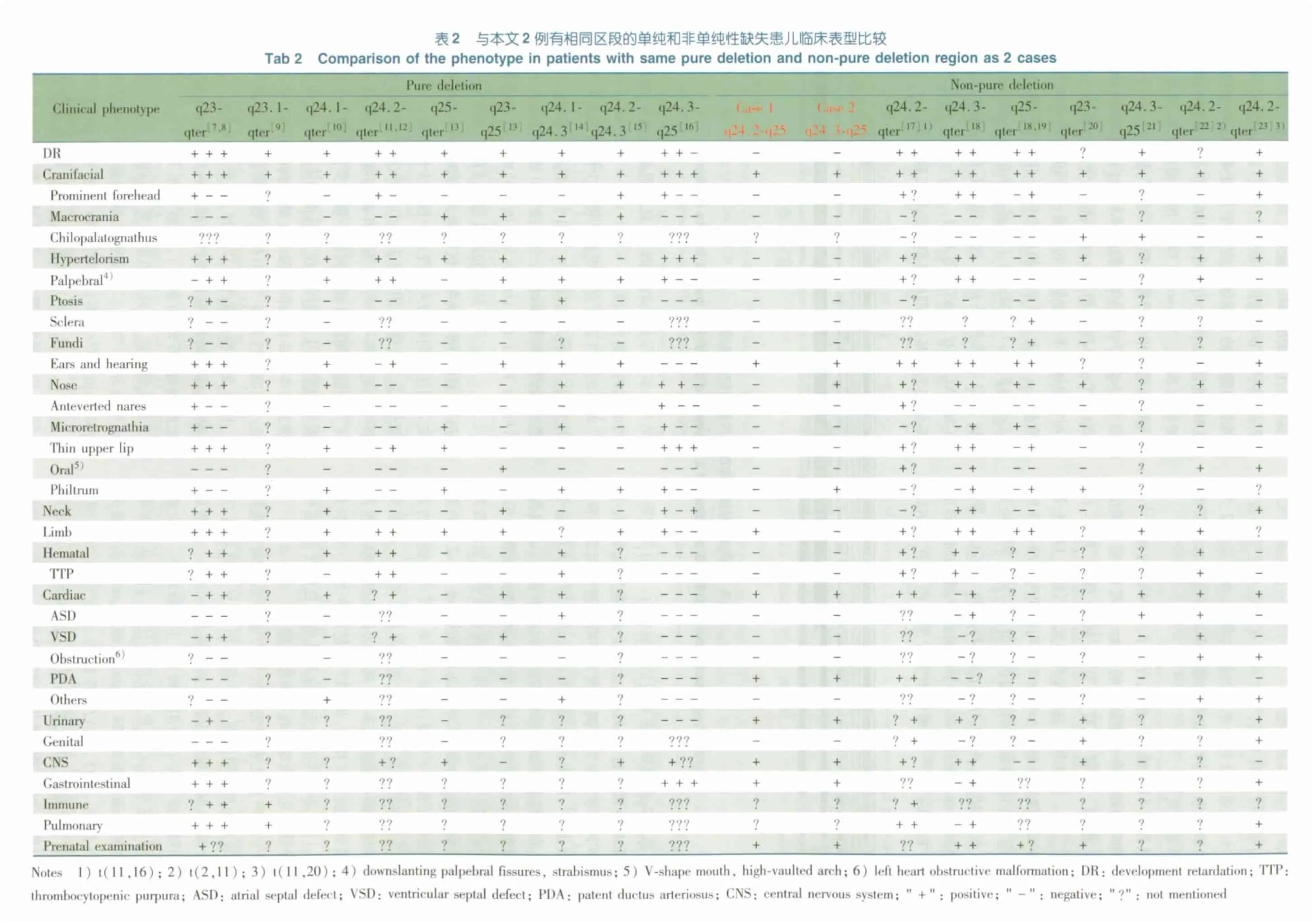
以11 号染色体长臂的缺失、短臂的重复和Jacobsen 综合征为检索词,检索PubMed 数据库和中国知网,检索截至时间为2005 年1 月至2012 年1 月。表2 显示,检索到与本文2 例有相同区段的单纯性缺失患儿14 例[7~16],9 个区段;非单纯性缺失患儿10 例[17~23],7 个区段,本文2 例为非单纯性缺失患儿。表2 报道的单纯性缺失患儿涉及的临床表型包括生长发育迟滞(13/14 例),头面部畸形(14/14例),血液系统异常(6/11 例)、心血管系统异常(6/11 例),头颅影像学异常(7/8 例);非单纯性缺失患儿涉及生长发育迟滞(8/8 例),头面部畸形(10/10 例),血液系统异常(3/6 例)、心血管系统异常(6/8 例)和头颅影像学异常(4/8)。例1 和2 除表现为与文献报道一致的JBS 关键区域的缺失外,相关的临床表型也较为相似。例1 表现为头面部畸形(低位耳),心血管系统异常(卵圆孔未闭、动脉导管未闭),泌尿生殖系统畸形(睾丸未降),神经系统异常(右侧尾状核头部、右侧脉络膜体部囊性占位)。例2 表现为头面部畸形(小鼻、短人中、舌喜外伸),心血管异常(动脉导管未闭、卵圆孔未闭),内脏畸形(脾肿大),神经系统异常(双侧脑室扩大、不对称、左侧脉络膜头部囊性占位和脑发育不全)。本文2 例均为生后1 ~2 d 的新生儿,自动出院后因家长不配合未能进一步随访,无法得知是否存在生长发育迟滞。
从表2 可见,文献[16]报道的3 例11q24.3-q25 的患儿与本文例2 缺失的区段相同,但临床表型存在差异。分析原因可能为:①JBS 存在异质性,相同基因型的个体表型存在差异[11,12];②既往报道大部分病例均为缺失的区段,很少准确的定位断裂的起始位点。所以相同区段的缺失,可能因为断裂位点的不同,导致其临床表型不完全一致。③文献[16]报道的3 例为单纯性缺失患儿,而本文例2 为非单纯性缺失患儿,例2 临床表型的不同可能与合并其他染色体异常相关。
?
文献报道51%的JBS 患儿存在头颅MRI 的异常[3]。已报道MRI 的异常有双侧脑室的扩大、不对称,脑白质的减少,脑萎缩,小脑发育不良和胼胝体发育不良等[7,8,11,16,17,24]。既往报道与脑发育相关的区段为11q24.2-q25,与本文2 例患儿相同。本文2 例均表现为头颅MRI 的异常,从图3 可见,在缺失的区段内报道的与神经系统相关的基因有 BARX2、ARHGAP32、APLP-2 和 B3GAT1 等。BARX2 基因含有4 个外显子,位于缺失区段,编码同源异形盒蛋白。Krasner 等[25]发现鼠类发育的神经元中,BARX2基因高表达。ARHGAP32 基因在脑内高表达,在神经元的发育过程中调节树突的形状和长度[26]。Grossfeld 等[3]将导致智力障碍的关键区域定位于接近端粒的一端长度为6.8 Mb 的区域,位于此区域的B3GAT1 基因是智力障碍的重要候选基因。Yamamoto 等[27]报道敲除此基因的小鼠,其突触可塑性和学习能力都会受到严重损害。APLP-2 基因位于11q24.3,编码类淀粉前体样蛋白,属淀粉前体蛋白家族(APP、APLP1、APLP2),在脑组织中广泛表达。APLP2与APP 协同调节神经肌肉的传递,空间的学习和突触的可塑性相关。本文2 例缺失的区段位于JBS 的关键区段,缺失大小为5.6 ~7.5 Mb,除了上述提到的在此缺失区段内与脑发育可能相关的候选基因,还有SNX19,THYN1,OPCML,NCAPD3 及NTM 等基因。迄今为止,报道的与神经系统相关的基因,都尚停留于动物实验阶段。
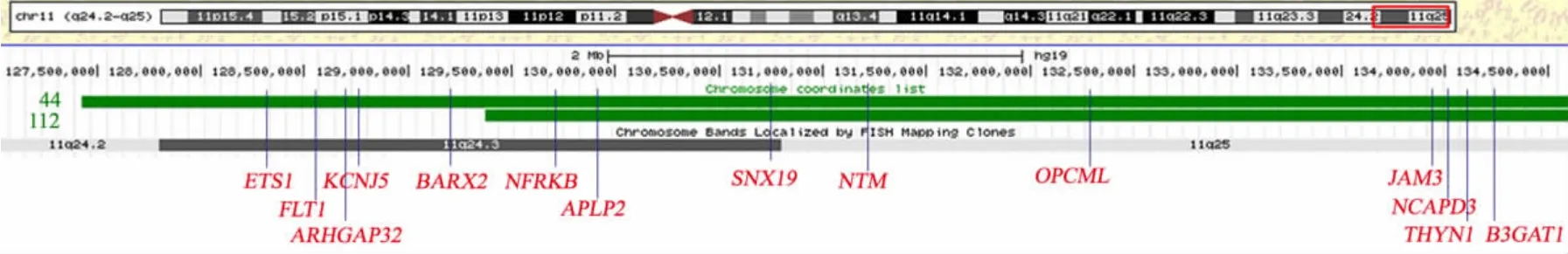
图3 2 例11 号染色体缺失片段所包含基因Fig 3 Regions of the 11q24-q25 deletion of 2 cases and candidate genes in this region
JBS 患儿头面部异常主要包括前额突出、三角头畸形、眼距宽、眼睑不能闭合、内眦皮、上睑下垂、鼻梁低平、人中异常和小下颌等。本文2 例表现为不典型的头面部异常。有研究报道[28],影响头面部发育的候选基因BARX2 位于11q24.3,在神经和颅面部结构的发育过程中表达。例1 缺失的区段包含这一基因,但是表现出不典型的头面部畸形;例2 缺失的区段不包含这一基因,也表现为不典型的面部畸形。分析可能的原因:①本文2 例均为出生1 ~2 d 的新生儿,面部特征表现不明显,但由于自动出院后未能随访面部特征;②头面部的发育是一个剂量累积的效应。决定头面部发育的基因可能涉及多个,典型的头面部畸形可能是多个基因效应的相互作用,BARX2 只是其中之一。③本文2 例除了11 号染色体的缺失还合并其他染色体异常,这可能也是导致患儿头面部表现不典型的原因。
56%的JBS 患儿表现为先天性心血管系统畸形或异常,主要表现为房间隔缺损、室间隔缺损、动脉导管未闭、左心梗阻性畸形、肺动脉硬化和双右心室流出道等。最常见的是室间隔缺损和左心梗阻性畸形(主动脉畸形、二尖瓣狭窄、主动脉缩窄、左心发育不全综合征)。目前认为影响心脏发育的基因位于11q24.2-qter 区域[3],本文2 例缺失的区域也包含JAM3、KCNJ5 等影响心脏发育的基因。JAM3 已被确认是与JBS 心脏表型相关的基因[29]。KCNJ5在心脏中高表达,在JBS 患儿中有缺失报道。此外,ROBO4也是与心脏畸形相关的基因,研究表明其与血管的形成及保持心室的完整性相关。
94%的JBS 患者会出现血小板减少性紫癜或血小板功能的失调[3]。研究发现11q 与血小板相关的关键区域位于靠近末端约为6.8 Mb 的一个区段[3]。该区段包含了ETS-1、NFRKB、FLI-1 和JAM3 等与血细胞的形成及功能相关的基因[3,30]。本文例1 缺失的区段包含ETS-1、NFRKB、FLI-1和JAM3 基因;例2 缺失的区段包含NFRKB 和JAM3 基因。本文2 例住院期间均未发现血小板减少性紫癜。分析原因可能是:①文献报道JBS 发生血小板减少性紫癜的年龄可从新生儿至中年[14,18,19,31],发病年龄跨度较大,仅提示2 例新生儿住院期间没有观察到血小板减少性紫癜。②影响血小板功能的基因可能同时位于其他的染色体上,即使文献报道的与本文2 例相同区段缺失的病例也有部分患儿不出现血小板减少性紫癜[16],具体机制仍需进一步的研究。
BWS 是一种过度生长综合征,主要表现为巨大儿,巨舌,脐疝,面部畸形,胚胎源性的腹部肿瘤(Wilms 瘤、肾上腺皮质肉瘤、肝母细胞瘤),肾脏畸形等。11p15 区段为BWS 的关键区段,其致病机制主要为该区段印迹基因甲基化水平的异常[32~34]。本文例2 核型分析的结果表现为11q24.3-qter(5.6 Mb)缺失,具体表现为头面部异常(小鼻、短人中、舌喜外伸),胸前见一赘生物,心血管系统异常,脐疝,内脏异常(脾大),头颅MRI 和B 超异常,听力损害(双耳重度听力损害)。一方面与JBS 相似,另一方面例2 重复片段也涉及11p15 (BWS 的关键区域),舌喜外伸可能与巨舌有关;脾肿大与BWS 的内脏肥大特征相似,其临床表型与BWS 有一定的相似度,可能与11p15 的大片段重复有关。
随着全基因组技术的飞速发展,尤其对于临床不易判断的非典型综合征,提供了更为准确的分子遗传信息。相对于高度异质性的表型,基因型的信息日趋完善。本研究报道的2 例仅依靠临床信息难以诊断的新生儿多发畸形患儿,全基因组拷贝数变异分析揭示了其复杂的染色体异常。虽然目前十分遗憾未能得到患儿家长配合,不能实现随访,使得部分表型的描述不够完整。但是,通过对于2 例共同缺失区段11q24-q25 与既往报道文献的分析比较,对于丰富这一区段临床表型信息具有重要意义,尤其针对罕见疾病,更多的相似报道的后续出现,才能使建立表型-基因型关联性成为可能。
[1]Mattina T,Perrotta CS,Grossfeld P. Jacobsen syndrome.Orphanet J Rare Dis,2009,4:9
[2]Jacobsen P,Hauge M,Henningsen K,et al. An (11;21)translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical,cytogenetical,and gene marker study. Hum Hered,1973,23(6):568-585
[3]Grossfeld PD,Mattina T,Lai Z,et al. The 11q terminal deletion disorder:a prospective study of 110 cases. Am J Med Genet A,2004,129A(1):51-61
[4]McPherson E,Meissner L. 11q-syndrome:review and report of two cases. Birth Defects Orig Artic Ser,1982,18(3B):295-300
[5]Wakazono A,Masuno M,Yamaguchi S,et al. Interstitial deletion of the long arm of chromosome 11:report of a case and review of the literature. Jpn J Hum Genet,1992,37(3):229-234
[6]Pivnick EK,Velagaleti GV,Wilroy RS,et al. Jacobsen syndrome:report of a patient with severe eye anomalies,growth hormone deficiency,and hypothyroidism associated with deletion 11 (q23q25)and review of 52 cases. J Med Genet,1996,33(9):772-778
[7]Boehm D,Laccone F,Burfeind P,et al. Prenatal diagnosis of a large de novo terminal deletion of chromosome 11q. Prenat Diagn,2006,26(3):286-290
[8]Afifi HH,Zaki MS,El-Gerzawy AM,et al. Distal 11q monosomy syndrome:a report of two Egyptian sibs with normal parental karyotypes confirmed by molecular cytogenetics. Genet Couns,2008,19(1):47-58
[9]Puglisi G,Netravali MA,MacGinnitie AJ,et al. 11q terminal deletion disorder and common variable immunodeficiency. Ann Allergy Asthma Immunol,2009,103(3):267-268
[10]Manolakos E,Orru S,Neroutsou R,et al. Detailed molecular and clinical investigation of a child with a partial deletion of chromosome 11 (Jacobsen syndrome). Mol Cytogenet,2009,2:26
[11]Bohm D,Hoffmann K,Laccone F,et al. Association of Jacobsen syndrome and bipolar affective disorder in a patient with a de novo 11q terminal deletion. Am J Med Genet A,2006,140(4):378-382
[12]Giampietro PF,Babu D,Zabel CA,et al. Novel clinical features in a child with partial deletion of chromosome 11[del(11)(q24.2)]:further evidence for phenotypic heterogeneity.Am J Med Genet A,2006,140(4):385-387
[13]Ji T,Wu Y,Wang H,et al. Diagnosis and fine mapping of a deletion in distal 11q in two Chinese patients with developmental delay. J Hum Genet,2010,55(8):486-489
[14]Wenger SL,Grossfeld PD,Siu BL,et al. Molecular characterization of an 11q interstitial deletion in a patient with the clinical features of Jacobsen syndrome. Am J Med Genet A,2006,140(7):704-708
[15]Tyson C,Qiao Y,Harvard C,et al. Submicroscopic deletions of 11q24-25 in individuals without Jacobsen syndrome:reexamination of the critical region by high-resolution array-CGH.Mol Cytogenet,2008,1:23
[16]Bernaciak J,Szczaluba K,Derwinska K,et al. Clinical and molecular-cytogenetic evaluation of a family with partial Jacobsen syndrome without thrombocytopenia caused by an approximately 5 Mb deletion del(11)(q24. 3). Am J Med Genet A,2008,146A(19):2449-2454
[17]Zahn S,Ehrbrecht A,Bosse K,et al. Further delineation of the phenotype maps for partial trisomy 16q24 and Jacobsen syndrome by a subtle familial translocation t(11;16)(q24.2;q24.1). Am J Med Genet A,2005,139(1):19-24
[18]Basinko A,Audebert-Bellanger S,Douet-Guilbert N,et al.Subtelomeric monosomy 11q and trisomy 16q in siblings and an unrelated child:molecular characterization of two der(11)t(11;16). Am J Med Genet A,2011,155A(9):2281-2287
[19]Evers C,Janssen JW,Jauch A,et al. A small terminal deletion 11q in a boy without Jacobsen syndrome:narrowing the critical region for the 11q Jacobsen syndrome phenotype. Am J Med Genet A,2012,158A(3):680-684
[20]Chen CP,Wang TH,Lin CC,et al. Prenatal diagnosis of partial trisomy 3p (3p21->pter)and partial monosomy 11q(11q23->qter)associated with abnormal sonographic findings of holoprosencephaly, orofacial clefts, pyelectasis and a unilateral duplex renal system. J Formos Med Assoc,2008,107(10):822-826
[21]Tan EC,Lim E,Cham B,et al. Partial trisomy 3p and partial monosomy 11q associated with atrial septal defect,cleft palate,and developmental delay:a case report. Cytogenet Genome Res,2011,134(4):319-324
[22]Podraza J,Fleenor J,Grossfeld P. An 11q terminal deletion and tetralogy of Fallot. Am J Med Genet A,2007,143A(10):1126-1128
[23]Courtens W,Wauters J,Wojciechowski M,et al. A de novo subtelomeric monosomy 11q (11q24. 2-qter)and trisomy 20q(20q13.3-qter)in a girl with findings compatible with Jacobsen syndrome:case report and review. Clin Dysmorphol,2007,16(4):231-239
[24]Gadzicki D,Baumer A,Wey E,et al. Jacobsen syndrome and Beckwith-Wiedemann syndrome caused by a parental pericentric inversion inv(11)(p15q24). Ann Hum Genet,2006,70(Pt 6):958-964
[25]Krasner A,Wallace L,Thiagalingam A,et al. Cloning and chromosomal localization of the human BARX2 homeobox protein gene. Gene,2000,250(1-2):171-180
[26]Okabe T,Nakamura T,Nishimura YN,et al. RICS,a novel GTPase-activating protein for Cdc42 and Rac1,is involved in the beta-catenin-N-cadherin and N-methyl-D-aspartate receptor signaling. J Biol Chem,2003,278(11):9920-9927
[27]Yamamoto S,Oka S,Inoue M,et al. Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem,2002,277(30):27227-27231
[28]Penny LA,Dell'Aquila M,Jones MC,et al. Clinical and molecular characterization of patients with distal 11q deletions.Am J Hum Genet,1995,56(3):676-683
[29]Phillips HM,Renforth GL,Spalluto C,et al. Narrowing the critical region within 11q24-qter for hypoplastic left heart and identification of a candidate gene,JAM3,expressed during cardiogenesis. Genomics,2002,79(4):475-478
[30]Santoso S,Sachs UJ,Kroll H,et al. The junctional adhesion molecule 3 (JAM-3)on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med,2002,196(5):679-691
[31]Choufani S,Shuman C,Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet,2010,154C(3):343-354
[32]Binder G,Begemann M,Eggermann T,et al. Silver-Russell syndrome. Best Pract Res Clin Endocrinol Metab,2011,25(1):153-160
[33]Demars J,Le Bouc Y,El-Osta A,et al. Epigenetic and genetic mechanisms of abnormal 11p15 genomic imprinting in Silver-Russell and Beckwith-Wiedemann syndromes. Curr Med Chem,2011,18(12):1740-1750
猜你喜欢
湘潮(上半月)(2022年8期)2022-12-12
中国典型病例大全(2022年7期)2022-04-22
健康体检与管理(2022年2期)2022-04-15
云南画报(2021年11期)2022-01-18
铁道通信信号(2021年6期)2021-07-08
科技视界(2020年8期)2020-05-18
铁道通信信号(2019年3期)2019-04-25
大观(2018年8期)2018-01-23
扬子江(2016年1期)2016-05-19
中国医疗美容(2015年1期)2015-07-12
