
烟酸在铁钝化膜层表面的吸附机理
2012-12-05 02:27田惠文李伟华王大鹏侯保荣
物理化学学报 2012年1期
田惠文 李伟华 王大鹏 侯保荣
(1中国科学院海洋研究所,山东省腐蚀科学重点实验室,山东青岛266071;2中国科学院研究生院,北京100049; 3青岛科技大学,教育部及山东省橡塑重点实验室,山东青岛266042)
烟酸在铁钝化膜层表面的吸附机理
田惠文1,2李伟华1,*王大鹏3侯保荣1
(1中国科学院海洋研究所,山东省腐蚀科学重点实验室,山东青岛266071;2中国科学院研究生院,北京100049;3青岛科技大学,教育部及山东省橡塑重点实验室,山东青岛266042)
循环伏安法结合原位拉曼光谱的表征结果表明,烟酸在铁钝化膜层表面的吸附行为归因于其具有形成稳定膜层复合物的性能,烟酸将间隙离子FeII转化为稳定的晶格离子FeIII,从而降低铁钝化膜的溶解性.旋转电极电化学晶体微天平的分析结果表明烟酸在活化态和钝化态铁表面的吸附行为遵循Langmuir-Freundlich热力学规律,并由此计算出过程中的吸附常数、标准自由能和非均质分布常数.研究认为有机分子在钝化膜表面为化学型吸附,可导致钝化膜破坏的间隙离子被烟酸固定在八面体空位晶格中形成稳定晶型结构,并通过扫描电镜(SEM)和衰减全反射红外光谱(ATR FTIR)分析对结论进行了再次验证.
烟酸;铁钝化膜;碱性介质;吸附
1 Introduction
Steel embedded in concrete is normally in a passive state against corrosion due to a thin iron oxide layer that forms on steel surface and remains stable in the high alkaline environment of the concrete.For the initiation of corrosion,this protective film can be destroyed(i.e.,de-passivation)and this can be mainly done by the presence of chlorides or by carbonation of the cover concrete.1,2The use of corrosion inhibiting admixtures has grown over the last 25 years because they provide protection and longevity to some extent that would be too expensive to achieve otherwise.3,4
An ideal inhibitor would be a compound preventing corrosion without unfavourable effects on the properties of concrete and also without environmental hazards.Generally,the inhibitors used in concrete are inorganic compounds such as phosphates,chromates,and nitrites5-7or organic compounds based on amines or organic acids,8-11although some of them have toxic effects and pollute the environment.Application of small bio-molecules(vitamin or antibacterial drugs often considered as possible substitutes)as environmental-friendly,low cost, readily available corrosion inhibitors would be favourable.Nicotinic acid,ascorbic acid,and mimosa tannin have been investigated also for comparative purposes as some previous work have shown that these compounds used in cases that pitting corrosion can act by competitive surface adsorption prior to the ingress of chlorides.12-14Ascorbic acid has already been proved as a good steel corrosion inhibitor in saturated Ca(OH)2solution.15-17
It is believed that the presence of an organic molecule in the corrosive media inhibits corrosion of metals by adsorbing at the metal-solution interface to suppress the aggressive ion incorporation.18-22The well known adsorption mechanism valid for the studies in oil and gas industry does not mean that it may be so successfully employed in reinforced concrete.23In the former case,the molecules act directly on bare steel in acidic and neutral media.However,due to the high alkalinity in concrete, there is already a protective oxide layer on the steel surface and the adsorption has to be on this passive layer rather than bare steel only.24,25Nowadays,very little information is available on the adsorption modes of this organic molecular-passive iron surface system.The interaction between the passive film and inhibitors is rarely reported.
In the previous work of our laboratory,nicotinic acid inhibitory properties towards steel corrosion were investigated in acidic media.26We also compared the nicotinic acid inhibitory action on different hot dipping coated steel substrates with the activity of two other environment-friendly inhibitors(acridine and berberine).27Rationalization of its superior performance was supported by the molecular modelling results.The aim of this work is to make an investigation on the adsorption mechanism of nicotinic acid on passive iron surface by means of cyclic voltammetry,in situ Raman and ATR FTIR spectroscopies, rEQCM tests,and SEM analysis.Yet there have been a number of questions that adsorption mode maybe also dependent on the chemical composition of aggressive ions and cement paste, but understanding how they function is an ongoing process that relies on experience to identify the most important issue(molecular and passive layer first)and on theory to untangle and describe the individual effects.
2 Experimental
2.1 Cyclic voltammetry and in situ Raman measurements
A self-designed Teflon cell was used for the cyclic voltammetry and in situ Raman investigations as shown in Fig.1.A 5 mm thick disc was cut from a 1 cm diameter iron rod(99.99%, Goodfellow Cambridge Limited)and encased in epoxy with only the polished surface exposed.The disc was connected to the Parstat-2273(Princeton Applied Research,America)as the working electrode.The reference electrode was a saturated calomel electrode(SCE)with Luggin capillary.The counter electrode was a Pt ring disc.NaOH(analytically pure)solutions(1 mol·L-1)were prepared because the redox processes in the passive layer were well differentiated and it was not far from the alkalinity found in cement pastes from standard Portland cement(the upper limit corresponding to highly alkaline cements).28Furthermore,unlike the other alkaline media,NaOH was considered inert which allowed treating adsorption of nicotinic acid as a single component system.29,30
Raman measurements were performed using a Jobin Yvon T64000 Raman spectrometer equipped with notch filter to separate the emitting light and the Rayleigh component.The excitation light was the green Ar laser beam(514.5 nm).Excitation laser power on the samples was filtered to 0.65 mW to avoid thermal effects for the sensitive iron oxides.The water immersion microscope attachment was a 50×long working objective. The detector was an air-cooled charge-coupled device having holographic gratings with 600 grooves·mm-1.
2.2 Adsorption measurements with the rEQCM
This study was based on methods31and patents32obtained by Kern et al.The mechanical parts of the experimental devices were prepared and fabricated by Wang from Max-Planck Society in Germany and Wang from Lanxess Energizing Chemistry in Germany and CH Electrochemical Instruments in China.

Fig.1 Self-designed device for in situ Raman investigationsCE:counter electrode;WE:working electrode;RE:reference electrode
A Zahner potentiostat(IM6 Germany)was used,and a platinum wire served as a counter electrode.Two reference electrodes were employed:a Hg/HgO(1 mol·L-1KOH)and a saturated electrode(SCE).All potentials in this paper were referred to the SCE because this electrode is the most employed one for measurements on concrete rebar in the laboratory.The rEQCM electrode and its characterization were described elsewhere in detail.31,32The 10 MHz AT cut quartz crystals with a sensitivity factor of 0.217 Hz·cm2·ng-1were contained in exchangeable holders mounted on a rotating shaft.33,34The 400 nm thick Fe electrodes were prepared by electro-deposition on a 190 nm thick sputter deposited Au film with an adhesion layer of 30 nm Cr.
For adsorption studies in the active potential region,35,36the electrodes were immersed in 0.1 mol·L-1NaClO4solution(analytically pure)and a cathodic polarization at-1.5 V was applied,a plateau in the EQCM signal after an initial mass loss indicated the complete removal of the oxide.All solutions were adjusted to 25°C and to pH 8.6 with NaOH which is the isoelectric point of Fe2O3passive film.37Nicotinic acid(analytically pure)has a pK value of 4.75,hence,it exists as anions in neutral solutions.Perchlorate used as the supporting electrolyte showed no specific adsorption on iron and NaOH was considered inert which allowed us to treat adsorption of nicotinic acid as a single component system.29,30
For adsorption in the passive potential region,the electrodes were immersed in 1 mol·L-1NaOH solution.The oxide was formed by sweeping the potential from-1.25 to 0.25 V at 5 mV·s-1and holding the potential at 0.25 V until current density had dropped to a few μA·cm-2and the frequency became stable.
The electrodes were polarized at-1.25 V for adsorption in the active region and at 0.25 V for the passive region and maintained at those potentials throughout the sequential nicotinic acid additions.35After applying a constant potential,the nicotinic acid concentration was increased in steps by adding 20 mL of solution containing 1×10-4,1×10-3(two times),5×10-3(two times),1×10-2(five times),and 2×10-2mol·L-1(five times)nicotinic acid to an initial volume of 50 mL solution.
The frequency change Δf is given with respect to the absolute resonant frequency of the quartz crystal and contains two contributions:38

where Δfm,adsis the frequency shift due to the adsorbing species at the electrode and Δfηis the concentration dependent frequency shift resulting from viscosity and density changes in the electrolyte layer sensed by the EQCM.However,effects of Δfηon the EQCM become important only at concentrations above 1×10-2mol·L-1,therefore,in the following,Δf always refers to the frequency values Δfm,ads.
2.3 SEM analysis
Cyclic voltammetry was carried out after cathodic reduction of the native films at-1.4 V for 1 min,scanning the potential from-1.25 to 0.25 V.In order to generate film thick enough to be characterised,each sample was cycled ten times at 1 mV·s-1.The solutions were 1 mol·L-1NaOH with addition of nicotinic acid with different concentrations(0,1×10-3,2×10-3, and 5×10-3mol·L-1).The surface morphologies of specimens after cyclic voltammetry in the absence and presence of nicotinic acid was observed on a KYKY2800B scanning electronic microscope.The accelerating voltage was 25 kV.
3 Results and discussion
3.1 Cyclic voltammetry analysis with in situ Raman and ATR FTIR spectroscopy
In order to investigate the adsorption mechanism of nicotinic acid onto iron surface,cyclic voltammetry in 1 mol·L-1NaOH blank solution and with addition of niacin with various concentrations were performed.The different peaks observed in voltammogram of Fig.2 are assigned as follows.39The peak I is attributed to the reduction of adsorbed hydrogen on the iron surface formed during the cathodic polarization.The anodic peaks II and III and cathodic peaks X and XI are assigned to the redox processes between Fe0and FeII:
It is possible that Fe0is derived from the disproportionation reaction of FeO in passive layer formed by the process associated to peak II,according to Eq.(3)

The presence of magnetite at highly cathodic potential corresponding to peak III was verified experimentally by in situ Raman spectra(Fig.3(A)).The transformation of FeIImay take place and the oxidation of FeIIIthus formed gives rise to peak III.
Peaks IV and IX are due to the formation of magnetite:

Peaks V and VI in anodic sweep and VII and VIII in cathodic sweep are attributed to the formation of two different FeIIIspecies obtained from magnetite oxidation:


Fig.2 Cyclic voltammogram of iron electrode in 1 mol·L-1NaOH blank solution purged with N2at a scan rate of 5 mV·s-1(1st cycle)
The formation of goethite needs less energy than maghemite because of topotactic nature of the reaction,thus may correspond to peaks V and VIII.The electric charge,Q(Fig.4),involved for each peak has been evaluated by considering a Gaussian function for convolution,in order to verify its compatibility with the reactions proposed above.39Fig.4 shows that the charges associated to peaks III-VI are close to that determined for the corresponding cathodic peaks VII-X.This observation corroborates therefore the association of different peaks made above.
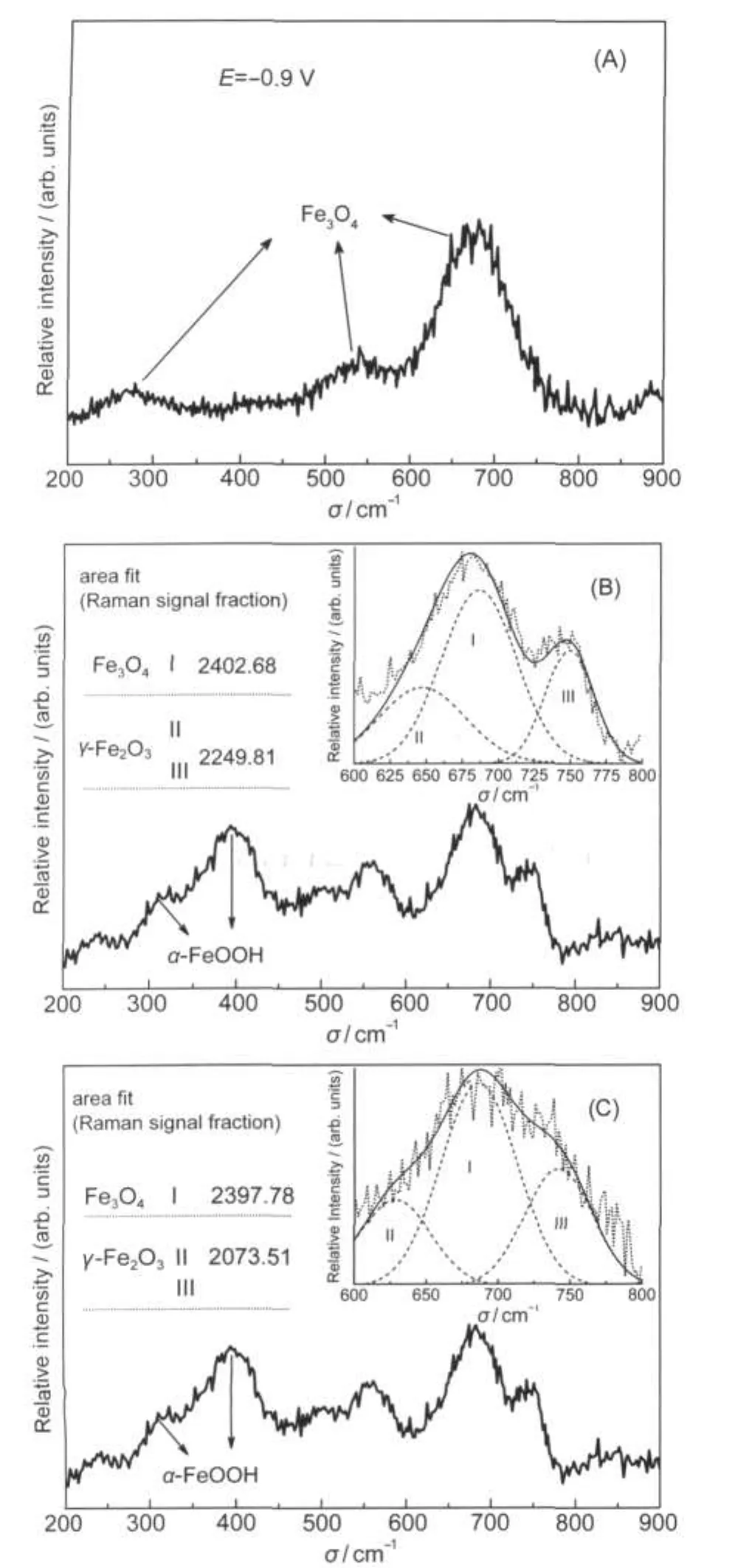
Fig.3 In situ Raman spectra of the iron electrode in 1 mol·L-1 NaOH blank solution at different anodic polarization potentials(A)E=-0.9 V;(B)E=-0.5 V;(C)E=0.5 V

Fig.4 Gaussian convolution and charge involved for each peak of the voltammogram given in Fig.2(A)anodic branch;(B)cathodic branch
It is postulated that there are two FeIIspecies having different energy states,one possibility is that anodic polarization from relatively negative values of around-1.1 V to more positive potentials,corresponding to the range of peak II,causes the formation of epitaxial FeIIin the oxide lattice.Another possible source is the interstitial FeII,dispersed in the oxide interstices,that continues forming in the whole forward scan interval of potential from-1.4 to 0.5 V.If FeIIIinvolved in peak III is entirely formed by the reaction depicted by Eq.(3),the charge of peak III should be one fourth of that of peak II.Fig.4 (A)indicates that this ratio is practically one half.This may indicate another source of FeIIthat transforms in this potential range to FeIII(or Fe3O4).Likewise,according to Eq.(4)the charge associated to peak IV should be one third of the sum of peaks II and III,whereas the observed value is much bigger. Both remarks probably corroborate the formation of interstitial FeIIwhen the potential scans positively from-1.4 to 0.5 V. This interstitial FeIIcould transform to FeIIIor Fe3O4and contribute to the process observed by anodic peaks.Certainly,further discussion is needed to prove this in the following sections.
According to Eqs.(5)and(6),the sum of charge involved in peaks V and VI is one third of that needed to completely transform magnetite formed at peak IV into ferric oxide.Fig.4(A) indicates that only one fifth of charge is found to form the latter species.That is to say,for the potential above peak IV,there is a high amount of accumulation of magnetite at the electrode surface.This consideration is supported by the results of Raman spectra(Fig.3(B,C))that indicate the presence of mag-netite in the potential sweep towards more anodic domain corresponding to a typical passive state.
When a real passivity scale is analyzed using Raman spectroscopy,it is seldom that one of the spectra obtained on pure iron oxide is recorded but often a mixture of different oxides present above.In order to correlate the Raman intensity with a mass fraction of each oxide,a decomposition method was therefore used to approximately quantify the amounts of different compounds.As revealed by Fig.3(B),the strongest peak at 675 cm-1proves the existence of magnetite,whereas,maghemite is characterised by the intensive bands at 650 and 750 cm-1.The formation of goethite is also evidenced by several narrow bands appeared in the range of 300-500 cm-1,which is consistent with Eqs.(5)and(6)proposed above.However,at potentials more anodic(Fig.3(C)),this species disappears probably because it dissolves.As already reported previously for the magnetite-maghemite binary mixture,the“shadowing effect”underlines that magnetite cannot be detected by Raman spectra unless it is present almost alone as pure compound.40,41Fig.3(B,C)shows that the contribution of magnetite to Raman signals remains a significant fraction of about 50%.40It can therefore be concluded that the passive layer experimentally detected is composed of a highly dominant magnetite and some other minor oxide compounds.
It is generally accepted that surface energy considerations can stabilize spinel Fe3O4over thermodynamically stable γ-Fe2O3,42,43which may help to explain why the magnetite is preferentially accumulated on the electrode surface rather than transformed into a hydrated FeIIIoxide.Thus,it could be deduced that only a small part of magnetite is used to the formation of FeIIIspecies,whose contribution to Raman signals is relatively weak(Fig.3(B,C))for a tiny mass fraction of less than 10%.40The maghemite in this potential range is therefore likely related to the electrical reorganisation of film with interstitial FeIII.The emission rate of this ion species that results from partial oxidation of interstitial FeIIas postulated above is not potential dependent.Coincidentally,Fe2+and Fe3+have been found leaving the ring-disk electrode at whole potential cycle,which indicates that a fraction of interstitial FeIIand FeIIIinject into the solution,and another trapped inside the film contribute to the process observed by anodic peaks.39

Fig.5 Schematic of physicochemical processes that occur with a passive film according to the truncated point defect modelFe=iron atom;Feiχ+=interstitial iron;Feδ+(aq)=iron cation in out layer/solution; FeFe=iron ion in cation lattice;VÖ=oxygen vacancy; OO=oxygen ion in anion lattice
The hypothesis upon which present work is based on is in good agreement with the point defect model.The truncated version of point defect model(PDM)as applied to iron in alkaline media is shown schematically in Fig.5.44,45Passive oxide film has been described as a bilayer consisting of an inner barrier layer based on magnetite-type structure,that grows directly on iron surface(reaction(C)),and an outer layer of γ-Fe2O3precipitated via the hydrolysis of interstitial iron cations ejected from the barrier layer(reaction(B))or by hydrolytic restructuring of the barrier layer itself.The film behaves as a highly doped n-type semiconductor due to a preponderance of iron interstitials and oxygen vacancies as electronic donors.
The above assumptions are confirmed by the analysis of cyclic voltammetry curves recorded in solutions containing nicotinic acid.First,as observed in Fig.6,the current densities decrease greatly as the compound is added into the solution,and the polarization curves move directly to lower current densities with increasing the concentration of nicotinic acid.However, addition of 5×10-3mol·L-1nicotinic acid does not reduce further current density obviously as compared with solution containing 1×10-3mol·L-1nicotinic acid.This phenomenon is in accordance with the previous work of our laboratory,26which showed that the corrosion of steel was evidently hindered even with lower nicotinic acid concentration,but inhibition efficiency increased slightly when the concentration was higher than 1×10-3mol·L-1.Further study is needed to find out an optimum dosage in the film forming process by rEQCM in the following sections.

Fig.6 Cyclic voltammogram of iron electrode in 1 mol·L-1NaOH solution with addition of various niacin concentrations
Second,the addition of nicotinic acid significantly alters the redox behaviour of passive film on iron.Under a positive polarization,two or three peaks are well resolved,corresponding to the successive oxidation of iron to higher valence state(Fe0→FeO→Fe3O4).Subsequently,in the backward scan,the overlapping reduction peaks appear,suggesting that a counterpart conversion of Fe3O4to Fe0.It is interesting to note that a significant current increase is observed for potentials more anodic than-0.5 V,and there is no corresponding cathodic reduction peak in the backward scan.This irreversible oxidation process may concern not only the formation of FeIIIspecies which is the important component of outer layer in PDM,but also the adsorbed nicotinic acid molecules,because it has been observed neither in Fig.2 nor in previous research works involving iron in NaOH solutions.46,47This argument is supported by ATR FTIR spectra obtained at the potential of peak VI up to-0.3 V.ATR FTIR spectra of the iron surface exposed in the solutions of various nicotinic acid concentrations as well as crystalline nicotinic acid were measured as shown in Fig.7. The bands with very low intensity are obtained for the electrode exposed to 5×10-4mol·L-1nicotinic acid solution,indicating smaller amount of adsorbed species.The majority of the bands observed in the spectrum of the electrode exposed to nicotinic acid solution of concentration up to 5×10-3mol·L-1, closely resemble those appearing in the crystalline nicotinic acid spectra.The disappearance of OH stretching vibrations would be related to the participation of iron-nicotinic acid bindings.The strong bands at 1700 cm-1in the reference spectrum, assigned to the C=O stretching vibration,has shifted to the lower wave numbers upon metal-ion binding.48,49The intense bands at around 1590 and 1480 cm-1are connected with C=N and C―N stretchings,neighbouring with the sharp adsorption at 1415 cm-1arising from C=C stretching of pyridine ring. Several bands at 1325-1035 cm-1observed in the crystalline spectrum,which are assigned to C―C(=O)―O stretching and the C―O―H bending modes,have suffered major loss in intensities in the spectrum of adsorbed organic molecules.This could be related to the C―O-group participation in the surface chelate formation.Furthermore,a new band with strong intensity occurs in the region 1060-1000 cm-1which is typical for the Fe―O―C group vibration.50Therefore,the disappearance of outer layer formation oxidation peaks can be interpreted in terms of reduced interstitial FeIIemission.In other words, organic molecules may act as depolariser of the out layer formation through the interaction with interstitial FeIIejected from the barrier layer.

Fig.7 ATR FTIR spectra of the crystalline nicotinic acid(A)and iron electrodes in 1 mol·L-1NaOH solution with addition of 5×10-3mol·L-1(B)and 5×10-4mol·L-1(C)nicotinic acid at anodic polarization potential of-0.3 V
Third,nicotinic acid chelating properties could account for both the irreversible oxidation process and the disappearance of out layer.Martinez15,16and Chang51et al.have proposed two mechanisms of iron oxide behaviour in the presence of a chelating agent:(1)solution coordination mechanism and(2)surface complexation mechanism.The first involves reaction of chelating agent with interstitial FeII,released from the barrier layer, via ligands thereby promoting autoxidation of FeIIto FeIII.The second assumes surface complexation of iron oxide with chelating species.In this case,after the formation of surface complex,relative bond strength of Fe-ion with lattice oxygen and with chelating molecule will be of outmost importance.If the lattice bond with Fe-ion is sufficiently weakened,the entire complex will be released into the solution.However,if the bond is stable enough,the complex will stay adsorbed at film surface,where the conformation of chelating agent in its adsorbed form will play an essential role.It was shown that this process is pH dependent and,in a higher pH environment,iron intersitials can be bonded to the terminal oxygen anions,giving iron-oxygen double bonds,at which the anion charge is delocalized and strong Lewis acidity can appear enabling formation of outer sphere complexes.Hence,interstitial FeIIrelease could be significantly reduced by formation of FeIII-complex that results in progressive development of surface insoluble film.Stabilization of iron in FeIIIstate can be explained by formation of highly stable surface complexes with the nicotinic acid ligands.Stability constants of the investigated compounds with FeIIIare listed elsewhere.52-54
Finally,Fig.8 shows that the charges involved in oxidation peaks II-V are four times that determined for reduction peaks. This discrepancy indicates that inner barrier layer is apparently stabilized by the adsorbed organic molecules such that the reduction reaction is suppressed in the cathodic scan.
3.2 Adsorption analysis with the rEQCM and SEM

Fig.8 Gaussian convolution and total charge involved for the peaks of the voltammogram given in Fig.6
Adsorption experiments with the stepwise addition of nicotinic acid were performed at active and passive iron surfaces and resulted in the frequency responses given in Fig.9.Adding the nicotinic acid to the base electrolyte led to an increase in resonance frequency,which according to Sauerbreyʹs equation55corresponds to a mass loss of the electrode.This behaviour is in qualitative agreement with the solvent substitution model proposed by taking into account the fact that the EQCM senses not only the mass of the electrode,but also that of the electrolyte layer immediately adjacent to the surface.35,56The or-ganic molecules adsorbing on the iron surface,occupying a large steric volume,replace several layers of much smaller but well-oriented water molecules.This leads to a decrease in the density of the electrolyte layer immediately adjacent to the electrode surface,and hence of the effective mass sensed by the crystal.36All Δf curves display a maximum with good reproducibility between different samples.It is assumed that this concentration corresponds to formation of a monolayer at the electrode.Apparently,more water is replaced during adsorption on iron than on iron oxide.The maximum is reached at lower concentration on active iron surface,which indicates an earlier monolayer formation than on passive iron surface.The adsorption amount is saturated on passive iron at the concentration of 5×10-3mol·L-1,and increases slightly when the added nicotinic acid concentration is higher than 1×10-3mol·L-1. This is in good agreement with the results obtained from cyclic voltammetry and our previous study.26Consistent with these observations,a XPS study showed that the inhibitor adsorbed better on iron in the active than in the passive state.57,58Linear sweep experiments also showed it to be more effective in suppressing the anodic dissolution in the active region.58This can be due to a different packing density of the organic molecules or to the nano-scale roughness and chemical effects of oxidized surface.On the other hand,the adsorption of nicotinic acid anion is rendered more easily since the iron surface is more positively charged with respect to the potential of zero charge. Therefore,the subsequent derivation of adsorption isotherms is needed to further elucidate the considerations.

Fig.9 Frequency shift of the nicotinic acid adsorption on active and passive iron as a function of nicotinic acid concentrationThe reproducibility of frequency response on passive iron is shown by short dot lines.
The surface coverage(θ)as a function of nicotinic acid concentration was obtained from EQCM data by the following equation:

The double logarithmic plots of θ/(1-θ)versus C were fitted to the Langmuir-Freundlich isotherm model(Eq.(7))and yielded straight lines with regression coefficients almost equal to 1 (Fig.10).


Fig.10 Curve fitting to the Langmuir-Freundlich isotherm for nicotinic acid adsorption on active and passive iron
where K is the adsorption constant,C the concentration of inhibitor,and h(0≤h≤1)the heterogeneity parameter.The LF model describes multisided adsorption on heterogeneous surfaces neglecting interactions between adsorbed molecules.
Adsorption constant K is related to the standard free energy of adsorption ΔG➝adsby Eq.(9).

All the obtained thermodynamic parameters are given in Table 1.The energy distribution of adsorption sites on active iron is clearly narrower than on oxide surface,indicating a more homogeneous behaviour.K andare significantly higher for passive film than for active iron.Generally,for values ofup to-20 kJ·mol-1,the type of adsorption is regarded as physisorption,the process is due to electrostatic interactions between moleculesand charged metal,while the values around-40 kJ·mol-1or higher are associated with chemisorption as a result of sharing or transferring of electrons to form coordinate type of bonds.Nicotinic acid adsorbed on iron in the active state forms bonds with adsorption energy comparable to complex interactions which may involve both chemisorption and physisorption.However,the value offor adsorption on iron in the passive state indicated that the mechanism is a typical chemisorption.
SEM photographs of the passive films obtained from iron surface after cyclic voltammetry in the 1 mol·L-1NaOH solutions in the absence and presence of 5×10-3mol·L-1nicotinic acid are shown in Fig.11.It can be observed that the passive film formed electrochemically on the iron surface in the absence of nicotinic acid is less homogeneous and reflects a loose growing pattern(Fig.11(A)).The original iron surface and scratches on the substrate induced by the emery paper are still partly visible.However,in the presence of nicotinic acid, the well differentiated morphology of Fig.11(B)reveals a ho-mogeneous and more compact passive film showing needle like shapes which is enough to cover the whole iron substrate. Davenport et al.43,59carried out an in situ surface X-ray diffraction study which demonstrated that the passive film formed electrochemically on oriented iron single crystals in a borate buffer solution had the spinel structure based on interstitial-excess Fe3O4.The spinel unit cell with a fully occupied 32 oxygen anions lattice contains 16 octahedral and 8 tetrahedral iron cation sites.In their ideal positions,they form a face-centred cubic(FCC)lattice(close-packed array).

Table 1 Fitting results from the Langmuir-Freundlich isotherm applied to rEQCM adsorption data

Fig.11 SEM photographs of the passive films obtained from iron surface after cyclic voltammetry in 1 mol·L-1NaOH solution and corresponding partial enlargement pictures in the absence(A)and presence(B)of 5×10-3mol·L-1nicotinic acid

Fig.12 Interstitial-excess spinel structure of the passive film formed on iron surface
Fig.12 shows the bottom half of one unit cell of the spinel structure model based on our own assumption with the z dimension.The upper half of the cell is identical to the lower half, but rotated by 90°.The structure of the passive film has octahedral site occupancy of 80%and tetrahedral site occupancy of 66%;there are cations occupying 12%of the available octahedral interstitial site,but no tetrahedral interstitials.In aqueous alkaline media,the nicotinic acid exists either as negatively charged species with respect to the point of zero charge(PZC) or in the form of anions which is stabilized by intramolecular hydrogen bond.60It is reasonable to propose that the adsorption mechanism on passive film involves reaction of nicotinic acid with octahedral interstitial iron cations dispersed in the crystallite,as shown in Fig.12,via ligands thereby promoting autoxidation of FeIIto FeIII.The considerations leading to this assumption have been proved by the results obtained from cyclic voltammetry and in situ Raman spectroscopy investigations. After the formation of complex,organic molecules attached to film surface by means of chemisorption,where the interstitial cations were fixed in the octahedral sites for a stable nano-crystalline and more compact film.It is likely that there is a correlation between the tetrahedral vacancies and the octahedral interstitials:these interstitial cations have a high probability of occupying sites near tetrahedral vacancies due to considerable electrostatic repulsion.Therefore,an inhomogeneous depth distribution of cations cannot be excluded.
4 Conclusions
Nicotinic acid significantly altered the redox behavior of the passive oxide film.The interstitial iron cation emission could be reduced by formation of FeIII-complex.Stabilization of iron in FeIIIstate can be explained by formation of highly stable surface complexes with the nicotinic acid ligands.
The adsorption model of nicotinic acid on iron in the active and passive states obeyed Langmuir-Freundlich isotherm.The adsorption process was spontaneous.The value of the standard free energies of adsorption indicated that adsorbtion of nicotinic acid on iron in passive state was typical chemisorption mechanism.
ATR-FTIR allowed concluding that organic molecules attached to passive film surface by means of chemical adsorption,where the interstitial cations were fixed in the octahedral sites for a stable nano-crystalline to enhance the corrosion resistance of the film.
(1) Söylev,T.A.;Richardson,M.G.Constr.Build.Mater.2008,22, 609.
(2) Jussain,R.;Ishida,T.Constr.Build.Mater.2011,25,1305.
(3) Chen,W.;Du,R.;Ye,C.Electrochim.Acta 2010,55,5677.
(4) Wei,J.;Dong,J.;Ke,W.Constr.Build.Mater.2011,25,1243.
(5)Ann,K.J.;Jung,H.S.;Kim,H.S.Cem.Concr.Res.2006,36, 530.
(6) Garcés,P.;Saura,P.;Méndez,A.Corrosion Sci.2008,50,498.
(7) Sanchez,M.;Alonso,M.Constr.Build.Mater.2011,25,873.
(8)Al-Mehthel,M.;Dulaijan,S.Construc.Build.Mater.2009,23, 1768.
(9) Ormellese,M.;Lazzari,L.;Goidanich,S.;Fumagalli,G.; Brenna,A.Corrosion Sci.2009,51,2959.
(10) Jamil,H.E.;Shriri,A.;Boulif,R.Cem.Concr.Comp.2005,27, 671.
(11) Jamil,H.E.;Shriri,A.;Boulif,R.Electrochim.Acta 2004,49, 2753.
(12) Ferreira,E.S.;Giacomelli,C.;Spinelli,A.Mat.Chem.Phys. 2004,83,129.
(13) Goncalves,R.S.;Mello,L.D.Corrosion Sci.2001,43,457.
(14)Akrout,H.;Bousselmi,L.;Dalard,F.J.Mater.Sci.2004,39, 7341.
(15) Valek,L.;Martinez,S.;Brnardić,I.Corrosion Sci.2008,50, 2705.
(16) Martinez,S.;Valek,L.;Oslaković,I.S.J.Electrochem.Soc. 2007,11,C671.
(17) Valek,L.;Martinez,S.;Serdar,M.;Oslaković,I.S.Chem. Biochem.Eng.Q.2007,21,65.
(18) Soylev,T.;McNally,C.Cem.Concr.Res.2007,37,972.
(19) Soylev,T.;McNally,C.Cem.Concr.Res.2007,29,357.
(20) Trabanelli,G.;Monticelli,C.;Grassi,V.Cem.Concr.Res.2005, 35,1804.
(21)Mechmeche,L.B.;Dhouibi,L.;Zucchi,F.Cem.Concr. Compos.2008,30,167.
(22) Qu,Q.;Li,L.;Ding,Z.Corrosion Sci.2009,51,2423.
(23) Elsener,B.Corrosion Inhibitors for Steel in Concrete.In: International Congress of Advanced Materials,Their Processes and Applications,Munich,Germany,Aug 5-10,2000.
(24) Ormellese,M.;Taffaini,G.;Ganazzoli,F.Corrosion 2009,3,22.
(25)Diamanti,M.V.;Ormellese,M.;Pedeferri,M.Cem.Concr.Res. 2008,38,1349.
(26) Ju,H.;Li,Y.Corrosion Sci.2007,49,4185.
(27) Ju,H.;Kai,Z.;Li,Y.Corrosion Sci.2008,50,865.
(28) Taylor,H.Cement Chemistry,2nd ed.;T.Telford Pub.:London, 1998;p 214.
(29) Marinković,N.S.;Calvente,J.J.;Kováčová,Z.J.Electrochem. Soc.1996,143,L171.
(30) Ahlberg,E.;Friel,M.J.Electrochem.Soc.1990,137,1196.
(31) Kern,P.;Landolt,D.J.Electrochem.Soc.2000,147,318.
(32) Kern,P.;Agarwal,P.;Landolt,D.Design and Characterization ofARotating Electrochemical Quartz-crystal-microbalance Electrode.German Patent DE 19911291 C2,2001-09-21.
(33) Vatankhah,G.;Lessard,J.;Jerkiewicz,G.Electrochim.Acta 2003,48,1619.
(34) Méndez,A.;Diaz-Arista,P.;Trejo,G.Int.J.Electrochem.Sci. 2008,3,918.
(35) Kern,P.;Landolt,D.Electrochim.Acta 2001,47,589.
(36) Kern,P.;Landolt,D.J.Electrochem.Soc.2001,148,B228.
(37) McCafferty,E.J.Electrochem.Soc.1999,146,2863.
(38) Kern,P.;Landolt,D.J.Electroanal.Chem.2001,500,170.
(39) Joiret,S.;Keddam,M.;Takenouti,H.Cem.Concr.Compos. 2002,24,7.
(40) Dubois,F.;Mendibide,C.;Pagnier,T.Corrosion Sci.2008,50, 3401.
(41) Xiao,K.;Dong,C.;Li,X.J.Iron Steel Res.Int.2008,15,42.
(42) McHale,J.M.;Auroux,A.;Perrotta,A.J.Science 1997,277, 788.
(43) Davenport,A.J.;Oblonsky,L.J.;Ryan,M.P.J.Electrochem. Soc.2000,147,2162.
(44) MacDonald,D.D.;Sun,A.Electrochim.Acta 2006,51,1767.
(45)Zhang,Y.;MacDonald,D.D.;MacDonald,M.U.Corrosion Sci.2006,48,3812.
(46) Diáz,B.;Joiret,S.;Nóvoa,X.R.Electrochim.Acta 2004,49, 3039.
(47) Hamadou,L.;Kadri,A.;Benbrahim,N.Appl.Surf.Sci.2005, 252,1510.
(48) Tajmir-Riahi,H.A.J.Inorg.Biochem.1991,42,47.
(49) Tajmir-Riahi,H.A.J.Inorg.Biochem.1991,44,39.
(50) Paul,R.C.;Mohini,C.;Chadha,S.L.Transition Met.Chem. 1981,6,300.
(51) Chang,H.C.;Matijević,E.J.Colloid Interface Sci.1983,92, 479.
(52) Das,C.M.;Sudersanan,M.J.Appl.Electrochem.2003,33,333.
(53) Martell,A.E.;Smith,R.M.Critical Stability Constants; Plenum:London,1974.
(54) Martinez,S.;Stern,I.Chem.Biochem.Eng.Q.1999,13,191.
(55) Sauerbrey,G.Z.Phys.1959,155,206.
(56) Gan,F.;Dai,Z.;Wang,D.Corrosion Sci.2000,42,1379.
(57) Olsson,C.;Agarwal,P.;Landolt,D.Corrosion Sci.2000,42, 1197.
(58) Kern,P.;Landolt,D.Corrosion Sci.2002,44,1809.
(59) Toney,M.F.;Davenport,A.J.;Oblonsky,L.J.Phys.Rev.Lett. 1997,79,4282.
(60) Li,W.;He,Q.;Hou,B.Electrochim.Acta 2007,22,6386.
June 30,2011;Revised:October 11,2011;Published on Web:October 17,2011.
Adsorption Mechanism of Nicotinic Acid onto a Passive Iron Surface
TIAN Hui-Wen1,2LI Wei-Hua1,*WANG Da-Peng3HOU Bao-Rong1
(1Key Laboratory of Corrosion Science in Shandong,Institute of Oceanology,Chinese Academy of Sciences,Qingdao 266071, Shandong Province,P.R.China;2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China;3Key Laboratory of Rubber-Plastics of Ministry of Education and Shandong Province,Qingdao University of Science& Technology,Qingdao 266042,Shandong Province,P.R.China)
Cyclic voltammetry and in situ Raman spectroscopy were used to determine the adsorption mechanism of nicotinic acid onto passive iron film surface.Its ability to form a surface complex tends to stabilize the interstitial FeIIin FeIIIstates and results in the progressive development of an insoluble film. Furthermore,an analytical investigation using a rotating electrochemical quartz crystal microbalance (rEQCM)showed that the adsorption isotherms of nicotinic acid onto iron in the active and passive states followed Langmuir-Freundlich behavior from which the adsorption constant,standard free energy of adsorption,and heterogeneity could be calculated.The organic molecules attach to the film surface by chemisorption and the interstitial cations are fixed in the octahedral sites giving stable nanocrystals.These assumptions were confirmed by scanning electron microscopy(SEM)and attenuated total reflection transform infrared(ATR FTIR)spectroscopy.
Nicotinic acid;Passive film on iron;Alkaline medium;Adsorption
10.3866/PKU.WHXB201228137www.whxb.pku.edu.cn
*Corresponding author.Email:liweihua@qdio.ac.cn;Tel/Fax:+86-532-82898740.
The project was supported by the National Natural Science Foundation of China(51179182),Shinan Scientific and Technological R&D Foundation of Qingdao City,China(P2010-1-ZH-005),Ph.D.Foundation of Shandong Province,China(BS2009HZ002),and K.C.Wong Education Foundation,Hong Kong,China(20061231).
国家自然科学基金(51179182),山东省青岛市市南区科技发展基金(P2010-1-ZH-005),山东省博士后基金(BS2009HZ002)和香港王宽诚教育基金(20061231)资助项目
O646
猜你喜欢
保健医苑(2022年1期)2022-08-30
——李振声
干旱地区农业研究(2022年3期)2022-06-09
模具制造(2022年3期)2022-04-20
绿色建筑(2020年1期)2020-07-15
中山大学学报(自然科学版)(中英文)(2020年1期)2020-02-26
模具制造(2020年12期)2020-02-06
安徽化工(2019年2期)2019-06-03
上海包装(2019年2期)2019-05-20
中国科学院院刊(2016年8期)2016-03-24
糖尿病天地(临床)(2012年3期)2012-08-15
