
HPLC法测定乳癖散结颗粒中迷迭香酸的含量
2012-11-22 01:08王云霞王敏春谢志民西安市食品药品检验所西安710054
陕西中医 2012年4期
王云霞 王敏春 谢志民 西安市食品药品检验所(西安710054)
乳癖散结颗粒是国家食品药品监督管理局2008年批准生产的新剂型,由夏枯草、川芎、僵蚕、鳖甲、柴胡、赤芍、玫瑰花、莪术、当归、延胡索、牡蛎等11味药材组成,原标准无含量控制方法。夏枯草为方中君药,以往资料多采用高效液相色谱法、气相色谱法、薄层扫描法或毛细管电泳法测定其齐墩果酸或熊果酸的含量,作为其质量控制方法;近年来有以高效液相色谱法测定其迷迭香酸的含量,作为其质量控制方法[1-3],均使用十八烷基硅烷键合硅胶为填充剂。本文使用极性乙醚连接苯基键合硅胶柱,采用HPLC方法测定其所含迷迭香酸的含量,作为成方制剂的质量控制方法。
1 仪器与试药 1.1仪器 岛津LC-2010AHT型高效液相色谱仪、LC-solution色谱工作站,北京普析通用分析仪器有限责任公司TU-1800PC型紫外-可见分光光度计。
1.2 试剂与试药 迷迭香酸对照品由中国药品生物制品检定所提供(批号:111871-201001,供含量测定用),乙腈为色谱纯,其余试剂均为分析纯,水为双蒸水,乳癖散结颗粒样品为陕西白鹿制药股份有限公司提供,阴性样品、模拟低浓度样品、模拟高浓度样品均自制。
2 方法与结果 2.1 色谱条件与系统适用性试验 Phenomenex Synergi极性乙醚连接苯基键合硅胶柱(4μm,250mm×4.60mm),柱温30℃,检测波长为330nm,流速1.0mL/min-1,采用乙腈-0.1%磷酸梯度洗脱,时间程序见下表。
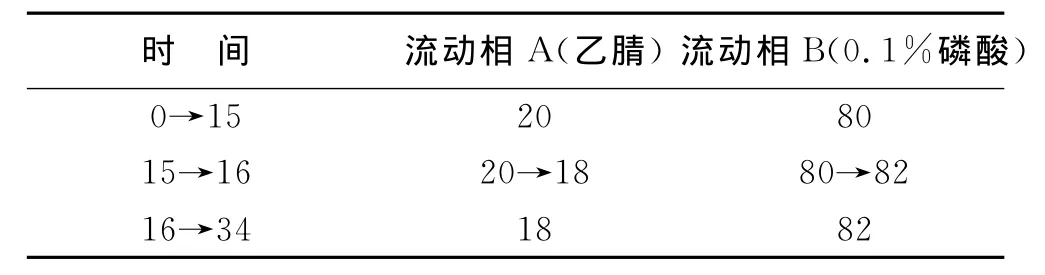
表1 时间程序表
在此条件下,样品得到良好分离,二极管阵列检测结果证明样品峰紫外吸收与对照品峰一致,夏枯草阴性对照样品在迷迭香酸峰处无峰出现,不干扰本品的测定。
2.2 对照品溶液的制备 精密称取迷迭香酸对照品10.01mg,置25mL量瓶中,加甲醇使溶解,并加至刻度,摇匀,作为6号对照品溶液,精密吸取6号对照品溶液1mL 5份,分别置5mL、10mL、25mL、50mL和100mL量瓶中,加乙腈-0.1%磷酸(20:80)的混合溶液至刻度,摇匀,分别作为5~1号对照品溶液。
2.3 供试品溶液制备 取重量差异项下的本品,研细(过3号筛),精密称取粉末2g,置具塞锥形瓶中,精密加入50%甲醇-甲酸(100:3.5)的混合溶液25mL,称定重量,加热回流1h,放冷,再称定重量,用上述混合溶液补足减失的重量,摇匀,置冰箱中冷藏2h,取出,滤过,弃去初滤液,取续滤液作为供试品溶液。
2.4 标准曲线与线性范围 按2.1中方法,精密吸取1~6号对照品溶液10μL(6号增加一15μL,1号增加一5μL),各进样2次,测定峰面积。以峰面积平均值为纵坐标,以进样量(μg)为横坐标,计算得回归方程 Y=3E+06X-7110.2,r=0.99992,表明在该系统条件下,迷迭香酸进样量在0.02ug~6.0ug范围内,峰面积积分值与进样量呈良好的线性关系。
2.5 精密度试验 精密吸取2.2项1、3、5号对照品10uL,各进样5次,按上述条件测定峰面积,结果RSD均小于1%。表明本方法进样量在0.04ug~0.8ug范围内进样精密度良好。
2.6 重复性试验 精密称取模拟1号样品(低浓度)、050828号样品和模拟2号样品(高浓度)2g,各3份,共9份,按照2.3项方法制备供试品溶液。精密吸取各供试品溶液和2.2项下2、3、5号对照品溶液各10μL,注入液相色谱仪,按2.1项下的条件测定迷迭香酸色谱峰面积,计算,结果表明样品每袋含迷迭香酸在0.45mg~3.96mg范围内,测定结果的RSD为0.2%~1.27%。
2.7 稳定性试验 取2.6中批号为050828的供试品溶液,按2.1项下方法,分别于0h、2h、4h、8h、12h、16h、20h和24h测定峰面积,结果峰面积RSD为0.43%,显示供试品溶液在24h内稳定。
2.8 回收率试验 精密称取迷迭香酸对照品12.02mg,置25mL量瓶中,加甲醇使溶解,并加至刻度,摇匀,作为Ⅰ号对照品溶液。精密吸取Ⅰ号对照品溶液3mL2份,分别置5mL和10mL量瓶中,加甲醇至刻度,摇匀,分别作为Ⅱ、Ⅲ号对照品溶液。分别精密称取模拟1号样品粉末、050828号样品粉末、模拟2号样品粉末1g,各3份,共9份,分别置具塞锥形瓶中。模拟1号样品分别精密加入III号对照品溶液1mL、050828号样品分别精密加入Ⅱ号对照品溶液1mL、模拟2号样品分别精密加入Ⅰ号对照品溶液2mL,分别精密加入50%甲醇-甲酸(100:3.5)溶液各25mL,按2.3中方法制备供试品溶液,以2.1中条件测定含量,计算回收率,结果(见表2)表明每袋含迷迭香酸在0.45mg~3.96mg范围内平均加样回收率为100.38%,RSD为1.35%~1.99%。
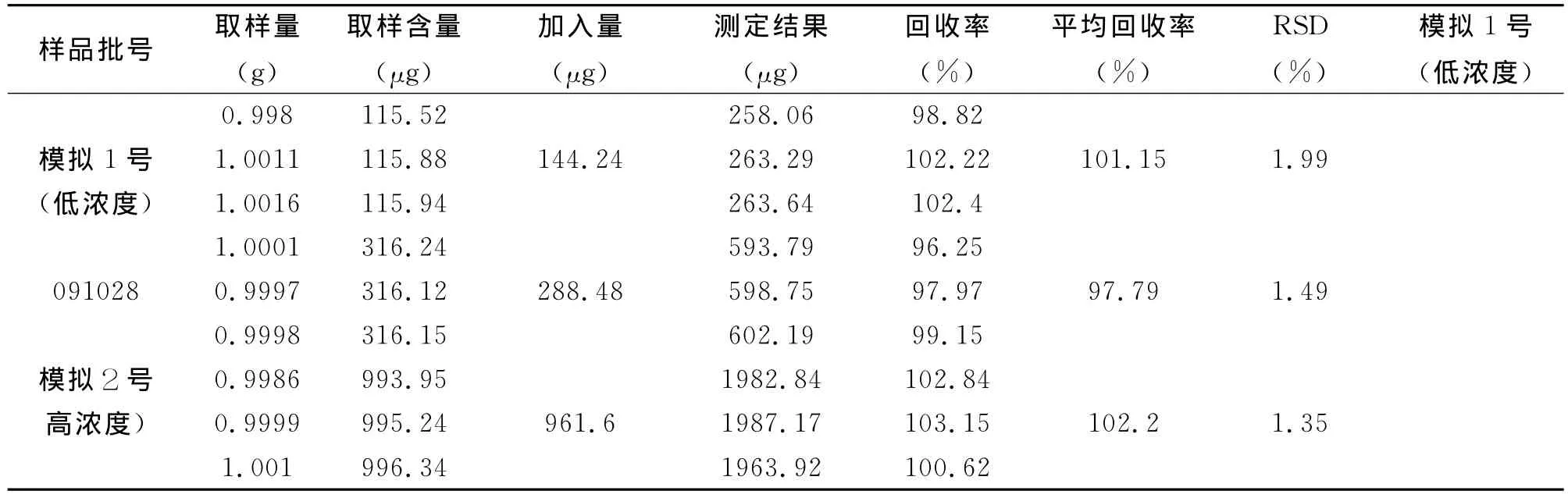
表2 迷迭香酸加样回收实验(n=9)
2.9 样品含量测定 精密称取6批样品各2g, 按2.3中方法制备溶液,以2.1中条件测定含量,结果6批样品含量为0.86mg/袋~1.71mg/袋(见表3),RSD为0.04%~1.04%。
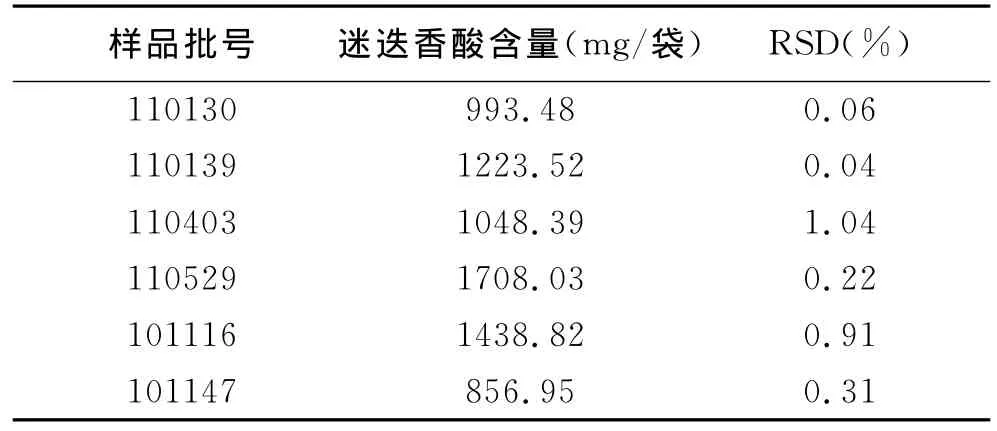
表3 各批次测定情况表
3 讨 论 3.1 研究过程中采用正交实验的方法对回流时间、甲酸的加入量等影响因素进行了考察 结果表明,回流时间和甲酸加入量对实验结果有显著影响,以50%甲醇-甲酸(100:3.5)为提取溶剂,回流1h为最佳。
3.2 样品溶液制备过程中发现 溶液放冷补重后直接滤过比较难,将此溶液置0~4℃冰箱中冷藏2h后较易滤过,不影响迷迭香酸的测定结果。
3.3 研究过程中同时对乳癖散结片和乳癖散结胶囊作了方法适用性研究 证明本法对乳癖散结片和乳癖散结胶囊中迷迭香酸的含量测定亦有良好的适用性。
3.4 系统耐用性试验 结果证明不同品牌的色谱柱、不同检测波长(326nm~334nm)、不同流动相比例(乙腈占18%~20%)、不同柱温(30℃~35℃)、不同流速(1.0mL/min~1.2mL/min)条件下迷迭香酸峰分离良好、峰形对称,均可用于迷迭香酸的含量测定。
[1]国家药 典委员会.中国药典[S].一部.北京:化学工业出版社,2010:197.
[2]王祝举,赵玉英,王 颁,等.夏枯草中迷迭香酸含量分析方法研究[J].药物分析杂志,2006,26(3):399.
[3]张兰珍,秦 雯,张小华,等.夏枯草不同部位中咖啡酸和迷迭香酸的含量测定方法研究[J].北京中医药大学学报,2007,5:9.
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09
家庭医学(2020年8期)2020-09-02
中国蜂业(2018年4期)2018-05-09
赤峰学院学报·自然科学版(2018年12期)2018-03-30
家庭医学(2017年9期)2017-10-24
中国民族医药杂志(2016年6期)2016-05-09
中国民族医药杂志(2016年4期)2016-05-09
当代化工研究(2016年6期)2016-03-20
中国卫生标准管理(2015年14期)2015-01-27
中国药业(2014年4期)2014-05-09
