
壬二酸固体分散体的制备及其表征
2012-11-22 05:31刘小平徐海星武汉理工大学化学工程学院制药工程系武汉430070
中国药房 2012年9期
金 晶,刘小平,徐海星(武汉理工大学化学工程学院制药工程系,武汉 430070)
壬二酸固体分散体的制备及其表征
金 晶*,刘小平#,徐海星(武汉理工大学化学工程学院制药工程系,武汉 430070)
目的:制备壬二酸固体分散体,改善壬二酸的溶出度,从而提高其生物利用度。方法:分别以聚乙二醇6000(PEG)、泊洛沙姆188为载体并选取药物与其不同比例(1∶3、1∶6、1∶9),采用熔融法、溶剂-熔融法制备壬二酸固体分散体,并对其进行体外溶出度的考察及比较;采用差示扫描量热法、X射线粉末衍射法鉴别壬二酸在固体分散体中的存在状态。结果:以PEG为载体的固体分散体的药物溶出优于以泊洛沙姆188为载体的固体分散体(90min内溶出分别为100%和80%);且当药物与PEG的比例为1∶9时,药物的溶出效果最好,与原料药比较药物溶出50%所需的时间大大缩短(12.65、45.65min)。壬二酸-PEG固体分散体中药物部分呈分子状态分散,部分呈微晶状态分散。结论:壬二酸与PEG(1∶9)的固体分散体能显著提高药物的溶出度。
壬二酸;固体分散体;聚乙二醇6000;泊洛沙姆188;体外溶出度;表征
壬二酸(Azelaic acid,AZA)是一种局部应用的非抗生素类治疗痤疮药物,具有长期用药后细菌不易产生耐药性、使用安全等优点[1]。目前已用于临床的壬二酸外用制剂有乳膏剂、凝胶剂和胶浆剂3种[2],但均存在一些不良反应,如局部灼痛、瘙痒、红斑等。引起这些不良反应的主要原因在于壬二酸的溶解性差,从而需要使用过量的壬二酸才能发挥治疗作用,而壬二酸的浓度越高,越容易对皮肤产生刺激[3]。因此,采用药剂学手段以增加壬二酸的溶解度,从而提高其生物利用度、减小其对皮肤的刺激,具有重要意义。固体分散技术是增加药物分散度、溶解度、溶出速率,提高药物生物利用度的一种有效方法[4]。本文以聚乙二醇6000(PEG)和泊洛沙姆188(PL 188)为载体,制备壬二酸固体分散体,对其体外溶出度进行研究,为其进一步的外用制剂研究奠定基础。
1 仪器与试药
1.1 仪器
1100型HPLC仪、二极管阵列检测器(DAD)(美国Agilent公司);RCZ-6C型药物溶出度仪(上海黄海药检仪器厂);STA449c/3/G型差示扫描量热仪(德国Netzsch公司);D/MAX-RB型X射线粉末衍射仪(日本Rigaku公司);FA2004N电子分析天平(上海精密科学仪器有限公司)。
1.2 试药
壬二酸对照品(德国Dr.Ehrenstorfer GmbH公司,批号:71001,纯度:99.0%);壬二酸原料药(医药级,四川西普化工股份有限公司,批号:20100514,纯度:98%);PEG(国药集团化学试剂有限公司);PL 188(北京偶合科技有限公司);其余试剂均为分析纯。
2 方法与结果
2.1 固体分散体的制备
2.1.1 壬二酸-PEG固体分散体的制备。
采用熔融法,按质量比1∶3、1∶6、1∶9准确称定壬二酸与PEG,先将载体在(80±2)℃水浴上加热熔融,再加入壬二酸,搅拌;使药物充分分散在载体中后,立即置于冰水浴上,剧烈搅拌,迅速冷却固化,再移至冰箱中冷冻2h后取出,置于40℃真空干燥箱中干燥24h,粉碎过筛,保存备用。
2.1.2 壬二酸-PL 188固体分散体的制备。
采用溶剂-熔融法,按质量比1∶3、1∶6、1∶9准确称定壬二酸与PL188,先将载体在(65±2)℃水浴上加热熔融,再加入以适量的无水乙醇溶解的壬二酸,不断搅拌,直至溶剂挥发完全,迅速移至冰箱中冷冻2h后取出,其余操作同“2.1.1”项。
2.1.3 物理混合物的制备。
按处方准确称取一定比例的PEG、PL188及壬二酸原料药,粉碎过筛,于乳钵中混匀,保存在干燥器中备用。
2.2 体外溶出研究
2.2.1 含量测定方法。
(1)色谱条件:色谱柱:ReproSil-Pur ODS(250mm×4.6mm,3.5µm);流动相:甲醇-磷酸二氢钾(pH=2.1)=55∶45,流速:0.8mL·min-1;柱温:30℃;检测波长:208nm;进样量:20µL。
(2)溶液制备。精密称取壬二酸对照品250.0mg置于100mL量瓶中,用流动相溶解并稀释至刻度,摇匀,得2.5mg·mL-1的对照品溶液。精密称取PEG 900.0mg置于50mL量瓶中,用流动相溶解并稀释至刻度,摇匀,得18.00mg·mL-1的PEG溶液;同法制备PL 188溶液。
精密称取壬二酸-PEG(1∶9)固体分散体500.0mg置于50mL量瓶中,用流动相溶解并稀释至刻度,摇匀,得壬二酸-PEG(1∶9)固体分散体供试品溶液。同法制备壬二酸-PL 188(1∶9)固体分散体供试品溶液。
(3)色谱行为。分别精密吸取对照品溶液、2种载体溶液、供试品溶液各20µL,进样分析。结果,壬二酸与溶剂峰分离良好,出峰处基线平稳无任何干扰,保留时间为9.3min,而载体材料均对药物峰无干扰,色谱详见图1。

图1 高效液相色谱图A.壬二酸对照品;B.PEG;C.壬二酸-PEG(1∶9)固体分散体;D.PL188;E.壬二酸-PL 188(1∶9)固体分散体;1.壬二酸Fig 1HPLC chromatogramsA.AZA control;B.PEG;C.AZA-PEG(1∶9)-SD;D.PL188;E.AZA-PL 188(1∶9)-SD;1.azelaic acid
(4)标准曲线。分别精密吸取对照品溶液0.2、0.6、1.0、2.0、4.0、6.0mL置于10mL量瓶中,用流动相溶解并稀释至刻度,摇匀,得浓度为0.05、0.15、0.25、0.5、1.0、1.5mg·mL-1的溶液,进样测定,以峰面积(A)对浓度(c,mg·mL-1)进行线性回归,得方程:A=678.75c+0.8824(r=0.9995)。结果表明,壬二酸检测浓度线性范围为0.05~1.5mg·mL-1。
(5)精密度。制备浓度分别为0.05、0.5、1.5mg·mL-1的壬二酸溶液,测定日内和日间精密度(n=3)。结果,低、中、高3个浓度的日内RSD分别为1.20%、0.32%、0.46%(<2%),日间RSD分别为0.60%、0.08%、0.26%(<2%),满足测定要求。
(6)回收率。按1∶9比例的处方量分别精密吸取一定量的对照品溶液和载体溶液(PEG/PL 188)置于量瓶中,用流动相稀释至刻度,摇匀,得壬二酸浓度为0.5、1.0、1.5mg·mL-1的溶液各3份,测定回收率。结果,PEG为载体的回收率试验中,平均回收率分别为98.59%、99.62%、100.36%,RSD分别为0.27%、0.09%、0.25%(<2%),满足测定要求;PL 188为载体的回收率试验中,平均回收率分别为98.98%、99.85%、100.30%,RSD分别为0.32%、0.41%、0.25%(<2%),满足测定要求。
2.2.2 体外溶出方法。
按《中国药典》2010年版附录ⅩC中篮法[5]规定进行,转速100r·min-1,水浴温度(37±0.5)℃,溶出介质(蒸馏)水900mL。精密称取壬二酸原料药、物理混合物及固体分散体(投药量相当于壬二酸0.9g),分别于10、20、30、45、60、75、90min各取样2mL,0.45µm微孔滤膜滤过,加流动相定容至5mL,摇匀,采用HPLC法测定含量。同时补充同温度的溶出介质2mL。将测得的结果代入标准曲线计算浓度,并换算成累积溶出百分率。
(1)药物与载体不同比例所制固体分散体的体外溶出情况。不同比例的壬二酸-PEG/PL 188固体分散体在水中的溶出曲线见图2。
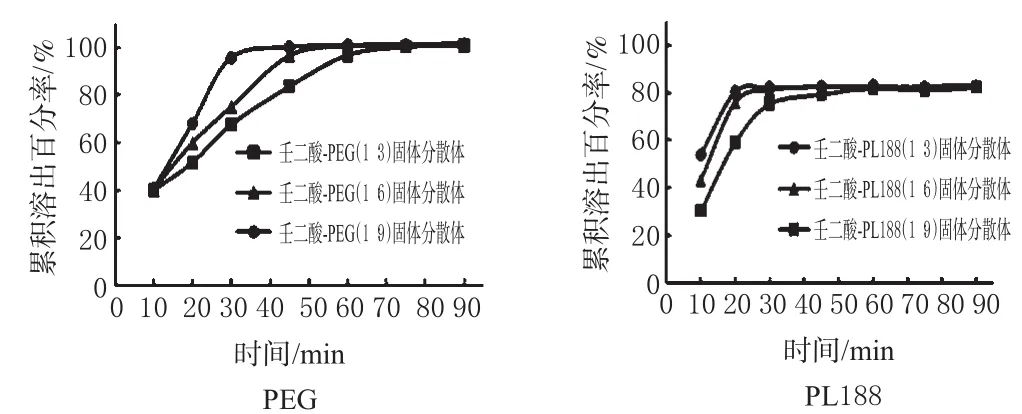
图2 药物与载体不同比例所制固体分散体的体外溶出曲线Fig 2 Dissolution profile of prepared solid dispersion with different drug-carrier ratio in vitro
从图2可看出,随着PEG用量增加,固体分散体中壬二酸的溶出速度增加;在90min内,不同比例的固体分散体中壬二酸的累积溶出百分率都能达到100%,即完全溶出。而随着PL 188用量的增加,固体分散体中壬二酸的溶出速度减小;在90min内,不同比例的固体分散体中壬二酸的累积溶出百分率只能达到80%,即没有完全溶出。
(2)溶出参数的统计分析。对上述2种固体分散体的溶出结果用Weibull分布模型拟合,求算溶出参数T50(药物溶出50%所需时间)和Td(药物溶出63.2%所需时间),结果见表1。
表1 2种固体分散体不同比例的溶出参数比较(Tab 1Comparison of dissolution parameter of 2kinds of solid dispersion with different drug-carrier rati(o

表1 2种固体分散体不同比例的溶出参数比较(Tab 1Comparison of dissolution parameter of 2kinds of solid dispersion with different drug-carrier rati(o
Td/min 23.0019.3715.9712.9116.4426.34壬二酸-PEG(1∶3)壬二酸-PEG(1∶6)壬二酸-PEG(1∶9)壬二酸-PL188(1∶3)壬二酸-PL188(1∶6)壬二酸-PL188(1∶9)16.1114.2512.658.0011.4817.31固体分散体T50/min
分别对2种固体分散体同种比例下的T50、Td进行t检验。结果显示,在3种比例下,2种固体分散体均具有显著性差异(P<0.05),再结合前文结果,认为壬二酸-PEG固体分散体的溶出效果较好。对2个比例(1∶3、1∶6)的壬二酸-PEG固体分散体的T50、Td与比例(1∶9)者的T50、Td进行t检验统计分析,结果显示,前二者与后者之间均具有显著性差异(P<0.05),故确定壬二酸-PEG固体分散体以1∶9组成最佳。
(3)壬二酸原料药、壬二酸-PEG(1∶9)的物理混合物、壬二酸-PEG(1∶9)固体分散体的体外溶出情况。三者在水中的溶出曲线见图3。
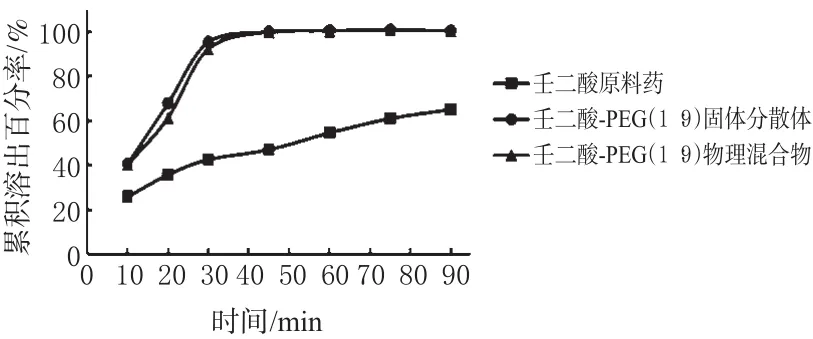
图3 3种样品的体外溶出曲线Fig 3 Dissolution profile of 3samples in vitro
从图3可看出,固体分散体的溶出速度和累积溶出百分率都明显高于原料药,但与物理混合物差别不大。
(4)溶出方程拟合。对上述原料药、物理混合物、固体分散体的溶出结果用Weibull分布模型拟合,求算T50和Td,结果见表2。
表2 3种样品的体外溶出参数结果Tab 2Dissolution parameter of 3samples in vitr(o3)

表2 3种样品的体外溶出参数结果Tab 2Dissolution parameter of 3samples in vitr(o3)
样品壬二酸原料药壬二酸-PEG(1∶9)物理混合物壬二酸-PEG(1∶9)固体分散体Td/min 87.5916.1515.97r 0.970.970.98T50/min 45.6512.9412.65
由表2可知,固体分散体的T50和Td最小,各自的相关系数r平均值>0.95,均满足要求,说明Weibull方程拟合的相关性较好。将固体分散体的T50、Td与原料药、物理混合物的T50、Td进行t检验统计分析,结果显示,固体分散体的T50、Td与原料药相比具有极显著性差异(P<0.01),与物理混合物相比T50、Td均无统计学差异(P>0.05)。
2.3 固体分散体的表征
2.3.1 差示扫描量热法[6]。
测试条件:以空铝干锅为参比,升温速率10℃·min-1,升温范围30~500℃,气氛为氮气。分别对壬二酸原料药、PEG、二者物理混合物(1∶9)及固体分散体(1∶9)进行差示扫描量热分析。结果见图4。
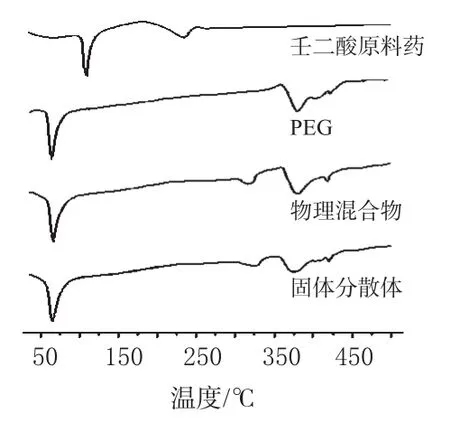
图4 4种样品差示扫描量热分析图Fig 4 DSC thermograms of 4samples

图5 4种样品X射线粉末衍射图Fig 5 X-ray powder diffraction patterns of 4samples
图4 结果表明,壬二酸原料药在108.4、234.7℃处有明显的吸热峰;PEG在65.9、355~448℃处有明显的吸热峰;固体分散体和物理混合物的图谱中壬二酸的吸热峰均消失。这说明固体分散体中药物可能以非晶体形式存在,并均匀分散在PEG中;而物理混合物则是由于在升温过程中,PEG先熔化并作为壬二酸的良好溶剂,且PEG比例较大,使壬二酸熔于其中,因而不显出壬二酸的吸热峰。
2.3.2 X射线粉末衍射分析[6]。
工作条件:Cu靶;高压强度:40kV;管电流:50mA;发散狭缝(DS):1°,散射狭缝(SS):1°,接收狭缝(RS):0.3mm;测速:15°·min-1;步宽:0.02°;扫描范围:5°~60°。分别对壬二酸原料药、PEG、二者物理混合物(1∶9)及固体分散体(1∶9)进行X射线粉末衍射分析,结果见图5。
图5结果表明,壬二酸在9.26°、18.68°、18.96°、22.80°处有特征衍射峰,且9.26°处的衍射峰特别强。PEG在19.28°、23.42°处有特征衍射峰。物理混合物的谱线中,壬二酸的特征峰减弱,只能看到9.26°处的特征峰,PEG的各特征峰明显存在;而在固体分散体的谱线中,壬二酸的特征峰几乎消失,无新的谱线出现。这说明无新的化合物产生,壬二酸在固体分散体中部分以分子状态分散,部分从固体溶液内析出呈微晶状态分散。
3 讨论
本文选择PEG和PL 188作为壬二酸固体分散体的载体材料,通过比较研究,发现PEG对壬二酸的体外溶出效果优于PL188,且当药物与PEG的比例为1∶9时,药物的溶出效果最好。
本试验选定的壬二酸-PEG(1∶9)固体分散体的累积溶出百分率与原料药间比较具有显著性差异,大大提高了壬二酸的溶出度。但有关将壬二酸-PEG固体分散体制备为凝胶及其生物利用度等情况有待进一步研究证实。
目前已上市的壬二酸软膏之所以容易对皮肤产生刺激,是因为在制备过程中将壬二酸细粉直接加入基质,故其中存在未溶解的壬二酸颗粒(晶体)。本试验制备的壬二酸-PEG固体分散体中,壬二酸一部分呈分子状态分散,一部分呈微晶状态分散,故能大大减小其对皮肤的刺激性。
[1] 陈冠容.壬二酸的药理作用和临床应用[J].中国医院药学杂志,2002,22(4):242.
[2] 张 宜,刘珍勇.壬二酸临床前药学研究进展[J].中国药业,2007,16(18):57.
[3]K·马瑟,C·R·斯托尔.含有溶剂化和稳定化壬二酸的局部用赋形剂[P].中国专利:CN1192134A,1998-09-02.
[4] 陆 彬.药物新剂型与新技术[M].北京:人民卫生出版社,2005:1-22.
[5] 国家药典委员会.中华人民共和国药典(二部)[S].2010年版.北京:中国医药科技出版社,2010:ⅩC.
[6] Manosroi J,Apriyani MG,Foe K,et al.Enhancement of the release of azelaic acid through the synthetic membrane by inclusion complex formation with hydroxypropyl-β-cyclodextrin[J].International Journal of Pharmaceutics,2005,293(1-2):235.
Preparation and Characterization of Azelaic Acid Solid Dispersion
JIN Jing,LIU Xiao-ping,XU Hai-xing(Dept.of Pharmaceutical Engineering,School of Chemical Engineering,Wuhan University of Technology,Wuhan 430070,China)
OBJECTIVE:To prepare Azelaic acid(AZA)solid dispersion(SD)and improve the dissolution of azelaic acid to promote the bioavailability of it.METHODS:AZA-SD was prepared using PEG6000(PEG),poloxamer188(PL188)as carriers by melting or solvent-melting methods with ratio of carriers to drug 1∶3,1∶6,1∶9.The dissolution in vitro was studied,and differential scanning calorimetry(DSC),X-ray powder diffraction were used to determine the status of azelaic acid in solid dispersion.RESULTS:The dissolution of AZA-SD with PEG as carrier was higher than AZA-SD with PL188as carrier,and their dissolutions were 100%and 80%within 90min.The dissolution of AZA-PEG(1∶9)-SD was the highest.50%raw material drug dissolved required 45.65min and that of AZA-PEG-SD required 12.65min.Drug in AZA-PEG-SD partly existed as molecule and partly as fine crystal.CONCLUSION:AZA-PEG(1∶9)-SD can improve the dissolution of azelaic acid.
Azelaic acid;Solid dispersion;PEG6000;Poloxamer188;Dissolution in vitro;Characterization
R943;R986
A
1001-0408(2012)09-0822-03
DOI 10.6039/j.issn.1001-0408.2012.09.19
*硕士研究生。研究方向:药物新剂型与新技术。E-mail:jinjing_janet@163.com
#通讯作者:教授。研究方向:药物新剂型与新技术、药物分析、药品质量标准。E-mail:pharmlxp@163.com
2011-04-02
2011-05-25)
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
承德医学院学报(2022年2期)2022-05-23
中国盐业(2018年20期)2019-01-14
新课程·下旬(2018年7期)2018-01-19
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
中成药(2017年6期)2017-06-13
中国当代医药(2015年33期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
