
可回收手性配体[QN(OH)2]2PHAL催化合成紫杉醇13-C侧链
2012-11-21 06:00南鹏娟赵海康崔新爱
合成化学 2012年3期
南鹏娟, 赵海康, 陈 晶, 崔新爱
(1. 陕西师范大学 材料科学与工程学院,陕西 西安 710062; 2. 西安医学院 第二附属医院,陕西 西安 710038)
紫杉醇(Taxol, Chart 1)是一种高效的细胞毒素[1],具有独特的抗癌机理、结构新颖、活性强、作用谱广。美国FDA在1992年将其作为抗晚期癌症的新药批准用于临床,目前已在全世界40多个国家上市,成为临床首选抗癌药物[2]。然而,紫杉醇的供应匮乏极大地限制了其临床应用。紫杉醇的直接来源是从几种红豆杉属植物树皮中提取分离,红豆杉生长缓慢,取树皮后可导致植物死亡,而且树皮中紫杉醇含量很低(约0.01%),从长远考虑根本无法满足日益增长的临床需要。虽然紫杉醇的全合成已获成功[3],但是全合成步骤长、产率极低,只有理论意义,而无实用价值。
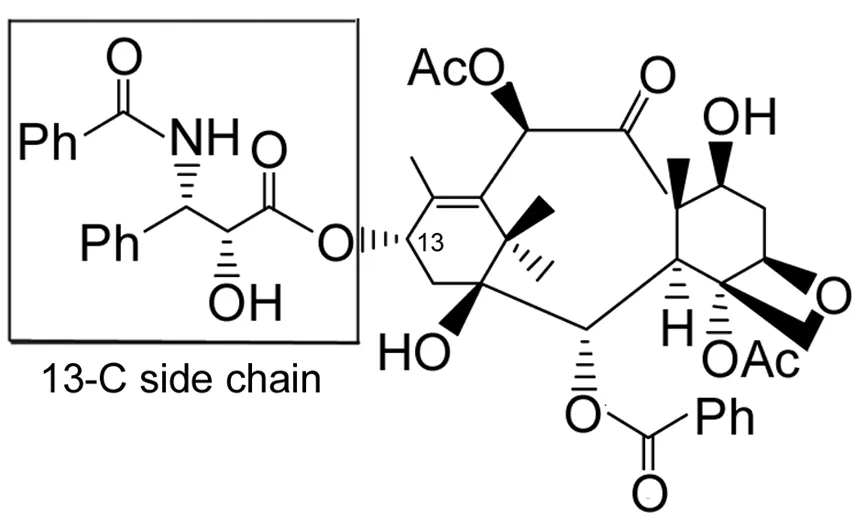
紫杉醇Chart 1
值得一提的是,红豆杉属植物树叶中富含10-去乙酰巴卡亭-Ⅲ(A, Chart 2),而且能够高效提取(1 g·kg-1新鲜树叶)。只要在A中引入N-苯甲酰基-(2R,3S)-3-苯基异丝氨酸侧链(13-C侧链),就可以半合成制得紫杉醇[4]。高效合成N-苯甲酰基-(2R,3S)-3-苯基异丝氨酸异丙酯(3)是半合成制得紫杉醇的关键。合成3的文献[5,6]方法很多,但大多存在产率低、反应步骤多、成本高等不足之处。简化3的合成方法是高产率制得紫杉醇的有效手段。
不对称双羟化(AD)反应和不对称氨羟化(AA)反应[7,8]在天然产物合成中的应用一直都在探索中,本文采用一种改进的方法合成了可回收的小分子手性配体[QN(OH)2]2PHAL(1, Chart 3);以1催化反式-肉桂酸异丙酯(4)通过AD反应或AA反应合成了3(Scheme 1),两种方法的产率分别为39%(97%ee)和52%(99%ee),其结构经1H NMR, IR和元素分析确证。
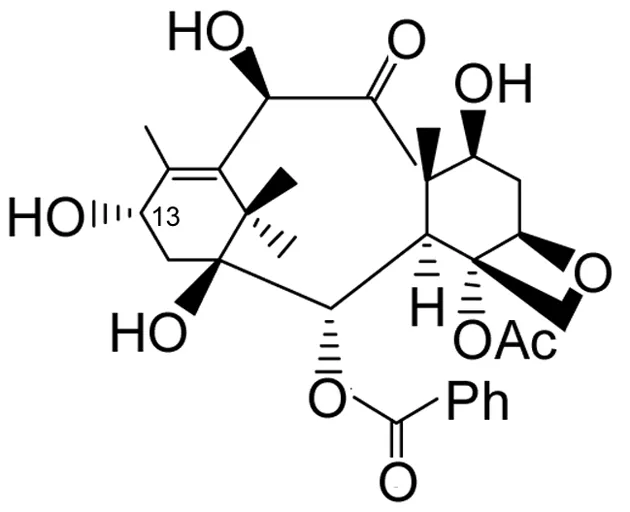
10-去乙酰巴卡亭-Ⅲ(A)Chart 2
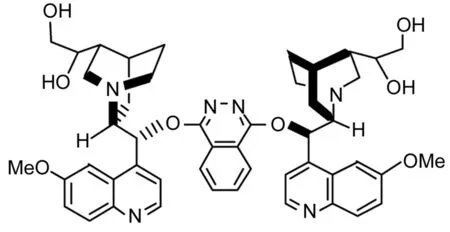
[QN(OH)2]2PHAL(1)Chart 3
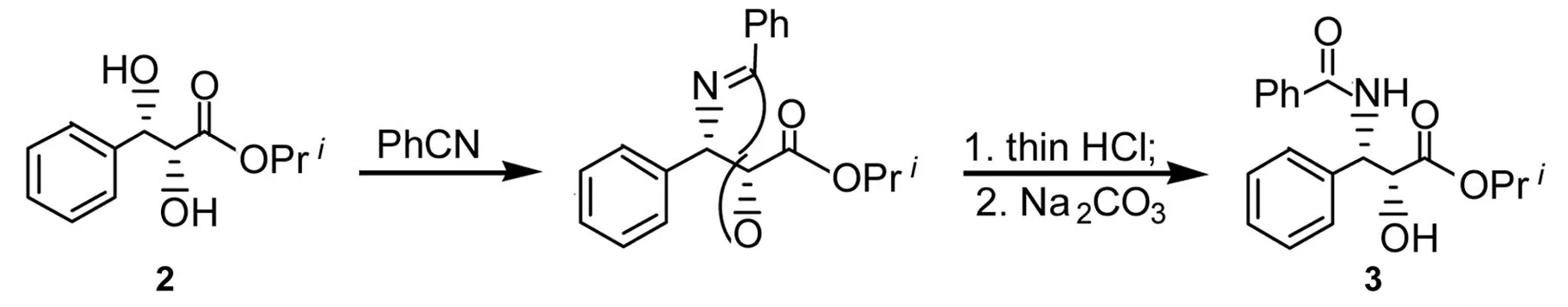
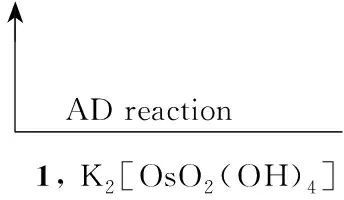
Scheme1
本文合成3所用的手性催化剂1可以回收和重复使用,极大地降低了成本。
1 实验部分
1.1 仪器与试剂
XRC-1显微熔点仪(温度未校正);PE343型自动旋光仪;Varian INOVA-400型核磁共振波谱仪(CDCl3为溶剂,TMS为内标);Waters-Breeze高效液相色谱仪(HPLC); VG ZAB-HS型质谱仪。
奎宁(QN),上海试剂二厂产品;氨基甲酸苄酯,Acros; CaH2, Merck-Schuchardt; 4, OsO4, K2[OsO2(OH)4], Aldrich; NMO(N-甲基-N-氧吗啉), ABCR; 1,4-二氯-2,3-二氮杂萘[9](m.p.162.0 ℃~163.5 ℃)和1[10](产率74%, m.p.199 ℃~201 ℃)参照文献方法合成;其余所用试剂均为分析纯。
1.2 合成
(1) AD反应合成3
在圆底烧瓶中依次加入丙酮90 mL,水10mL, OsO4260 μL(0.05mmol)的甲苯溶液(50 mg·mL-1)和1 0.45 g(1 mmol),搅拌10 min后加入NMO 1.75 g(1.5 mmol),剧烈搅拌下于0 ℃缓慢加入4 1.76 g(10 mmol)(约8 h),反应30 min。减压蒸除丙酮,用乙醚(3×50 mL)萃取(含1的水层留用),合并萃取液,用无水MgSO4干燥,减压蒸干,残余物用正己烷洗涤、干燥得(2R,3S)-(-)-2,3-二羟基-3-苯基丙酸异丙酯(2)。
在含1的留用水层中补加丙酮90 mL, OsO460 μL(0.01 mmol)的甲苯溶液(50 mg·mL-1)和NMO 1.75 g(15 mmol),剧烈搅拌下于0 ℃缓慢加入4 1.76 g(10 mmol)(约8 h),反应30 min。后处理同上。
同样的操作反复进行九次以上,所得2的平均产率90%, 99%ee。
(2) AA反应合成3
在圆底烧瓶中依次加入LiOH·H2O 403 mg(5.1 mmol)的水(15 mL)溶液和K2OsO2(OH)474 mg(0.2 mmol),搅拌下加入CH3CN 60 mL和10.9 g(1 mmol),搅拌10 min(溶液澄清)后加水15 mL,冷却至0 ℃,一次性加入4 0.85 g(5 mmol)和N-溴苯甲酰胺2.00 g(10 mmol),剧烈搅拌下于0 ℃反应至终点(TLC跟踪,反应液从深绿变成黄色),用Na2SO32.55 g终止反应。搅拌1 h,分液,水相用二氯甲烷(3×20 mL)萃取,合并有机相,用无水MgSO4干燥,浓缩至2 mL左右,搅拌下加入无水乙醚25 mL,析出白色沉淀,真空抽滤,滤饼即1(回收0.6 g);滤液减压浓缩后经硅胶柱层析[洗脱剂:V(乙酸乙酯) ∶V(正已烷)=1 ∶1]纯化得3 0.7 g,产率39%, 97%ee。其表征数据与AD反应合成的3一致。
2 结果与讨论
在众多金鸡纳生物碱衍生物中,以杂萘为桥连基的配体具有最优秀的化学活性和立体选择性,本文催化反应中所用到的1是以杂萘为桥连基的金鸡纳生物碱衍生物。关于1的合成,目前文献报道的方法有两种:第一种方法是Sharpless[11]报道的用K2CO3和KOH作缚酸剂,在溶剂甲苯中分水回流13 h制备。该方法合成温度高、反应时间长、产品需经柱层析纯化。第二种方法是以NaH作缚酸剂,DMF为溶剂,但是NaH的活性很强,在反应前须用无水乙醚处理,实验操作中NaH容易被空气氧化而着火,反应产物也需要经柱层析纯化,整个实验操作既繁琐又危险[12,13]。
在对金鸡纳衍生物的合成研究中,我们发现了一种新的缚酸剂CaH2[10]。 CaH2的活性低于NaH,投料前不需要作预处理,反应在1 h内可完成,反应的粗产物重结晶即可提纯。因此用CaH2作缚酸剂简化了1的合成方法,降低了成本。
Vorankov[14]于2003年报道了以4为底物经AD和Ritter反应合成3的方法。此方法反应步骤短、操作简单,但是第一步AD反应中用到的昂贵的配体却不能回收。我们在文献[14]方法的基础上作了三个改进:首先在第一步AD反应中采用了可回收的1作催化剂,并可以重复使用九次,2只需用正己烷洗涤即可纯化;其次将环化反应的时间缩短为3 h;最后采用沉淀析出的方法纯化3,避免了柱层析,降低了成本。
参考Song[15]用AA反应合成3的方法,改用可回收的1经一步反应合成3,产率39%, 97%ee;回收的1可直接重复使用,重复使用五次,催化活性未见明显下降。
[1] Kingston D G, Molinero A A, Rimoldi J M,et.al. Progress in the chemistry of organic natural products[M].New York:Springer-Verlag,1993.
[2] 徐铮奎. 植物抗癌药之王“紫杉醇”市场前景浅析[J].中国制药信息,2003,19(1):39-40.
[3] Denis J N, Greene A E, Gueard D. A highly efficient practical approach to naturaltaxol[J].J Am Chem Soc,1988,110(6):5917-5919.
[4] Nicolaou K C, Yang Z, Liu J J,etal. Total synthesis of taxol[J].Nature,1994,367:630.
[5] Commercon A, Bezard F, Bourzat J D. Improved protection and esterification of a precursor of the taxotere® and taxol side chains[J].Tetrahedron Lett,1992,33(36):5185-5188.
[6] Mukaiyama T. The unexpected and the unpredictable in organic synthesis[J].Tetrahedron,1999,55(29):8609-8670.
[7] Jacobsen E N, Marko I, Mungall W S,etal. Asymmetric dihydroxylation via ligand accelerated catalysis[J].J Am Chem Soc,1988,110(6):1968-1970.
[8] Li G G, Hubert H A, Sharpless K B.N-halocarbamate salts lead to more effieient catalytic symmetric aminohydroxylation[J].Angew Chem,Int Ed Engl,1996,35:2810-2813.
[9] Amberg W, Bennani Y L, Chadha R K,etal. Syntheses and crystal structures of the cinchona alkaloid derivatives used as ligands in the osmium-catalyzed asymmetric dihydroxylation of olefins[J].J Org Chem,1993,58(4):844-849.
[10] 南鹏娟,陈晶,孙晓莉.合成金鸡纳生物碱衍生物的简便方法[J].合成化学,2011,19(5):667-670.
[11] Sharpless K B, Amberg W, Bennani Y L. The osmium-catalyzed asymmetric dihydroxylation:A new ligand class and a process improvement[J].J Org Chem,1992,57:2768-2771.
[12] Jiang Ru, Kuang Y Q, Zhang S Y. A improved catalytie system for recycling OsO4and chiral ligand in the asymmetric dihydroxlation of olefins[J].Tetrahedron Asymmetry,2004,15(4):743-746.
[13] Wang Z M, Kolb H C, Sharpless K B. Large-scale and highly enantioselective synthesis of the taxol C-13 side chain through asymmetric dihydroxylation[J].J Org Chem,1994,59(17):5104-5105.
[14] Voronkovm V, Gontcharov A V, Wang Z M. Improved large-scale synthesis of phenylisoserine and the taxol C-13 side chain[J].Tetrahedron Lett,2003,44:407-409.
[15] Song C E, Roh E J. One-step synthesis of paclitaxel side-chain precursor:Benzamide-based asymmetric aminohydroxylation of isopropyl trans-cinnamate[J].Tetrahedron:Asym,1999,10(4):671-674.
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
世界农药(2019年2期)2019-07-13
吉林农业(2019年6期)2019-06-11
中成药(2017年10期)2017-11-16
实用口腔医学杂志(2017年6期)2017-09-19
中成药(2017年4期)2017-05-17
粘接(2017年4期)2017-04-25
中外医疗(2016年15期)2016-12-01
哈尔滨医药(2015年2期)2015-12-01
原子与分子物理学报(2015年3期)2015-11-24
