
PERK介导的内质网应激参与血管紧张素Ⅱ诱导心肌肥大的机制*
2012-11-06 08:31吕振嵘王晓礽李玉珍刘秀华
中国病理生理杂志 2012年7期
吕振嵘, 王晓礽, 李玉珍, 王 琛, 刘秀华△
(1山东大学医学院病理生理教研室, 山东 济南 250012;2中国人民解放军总医院病理生理研究室,北京 100853)
1000-4718(2012)07-1153-07
2012-03-05
2012-04-10
国家自然科学基金资助项目(No. 81070130)
△通讯作者 Tel: 010-66939763; E-mail: xiuhualiu98@yahoo.com.cn
▲并列第1作者
·论著·
PERK介导的内质网应激参与血管紧张素Ⅱ诱导心肌肥大的机制*
吕振嵘1▲, 王晓礽2▲, 李玉珍2, 王 琛2, 刘秀华2△
(1山东大学医学院病理生理教研室, 山东 济南 250012;2中国人民解放军总医院病理生理研究室,北京 100853)
目的研究蛋白激酶R样内质网激酶(PERK)介导的内质网应激(ERS)反应在血管紧张素Ⅱ(AngⅡ)诱导心肌细胞肥大中的作用。方法在AngⅡ诱导原代培养的乳大鼠心肌细胞肥大模型上,采用[3H]-亮氨酸掺入、心肌细胞表面积测定等评估心肌细胞肥大程度;以实时定量PCR、RT-PCR和Western blotting检测ERS标志性分子葡萄糖调节蛋白78(GRP78)、钙网蛋白(CRT)、蛋白激酶R样内质网激酶(PERK)、真核细胞翻译起始因子2α(eIF2α)和C/EBP同源蛋白(CHOP)mRNA和蛋白表达变化。结果与正常对照组比较,AngⅡ组CRT mRNA和蛋白表达分别高146.4%和125.3%(P<0.05),GRP78 mRNA和蛋白的表达分别高84.0%和77.6% (P<0.05),PERK mRNA和蛋白表达分别高165.4%和132.1%(P<0.05),eIF2α mRNA和蛋白表达分别高110.9%和46.5%(P<0.05),CHOP mRNA和蛋白表达分别高117.7%和63.3%(P<0.05)。结论PERK介导的内质网应激反应参与了AngⅡ诱导的心肌细胞肥大。
心肌肥大; 内质网应激; 蛋白激酶R样内质网激酶
心肌肥大是由缺血、机械牵张、激素和细胞因子等因素引起的心肌细胞代偿性反应,心肌肥大失代偿可导致扩张性心肌病、心力衰竭和猝死,严重危及人类生命。内质网(endoplasmic reticulum, ER)是真核细胞中调控蛋白质折叠、Ca2+稳态和应激反应的重要细胞器,对维持心肌细胞钙离子和蛋白质合成稳态具有重要作用[1]。缺血缺氧、葡萄糖/营养物质匮乏、ATP耗竭、大量自由基的产生及Ca2+稳态破坏等应激刺激均可引起内质网功能障碍,触发内质网应激(ER stress, ERS)。蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)是ERS 3种感受蛋白之一[2],通过磷酸化真核细胞起始因子2α抑制细胞蛋白质合成,并通过促进C/EBP同源蛋白(C/EBP homologous protein, CHOP)表达导致细胞凋亡,是介导ERS整合调控反应的重要细胞内信号途径之一[3]。本课题组前期研究证明[4],ERS诱导剂毒胡萝卜素(thapsigargin,TG;抑制ER钙泵继而排空ER内Ca2+)和衣霉素(tunicamycin,TM;抑制ER内蛋白质N端糖基化)可以直接诱导心肌细胞肥大。牛磺酸(taurine,Tau)是体内含量最丰富的含硫自由氨基酸,可以抑制同型半胱氨酸等多种因素诱导的ERS,对心肌细胞具有保护作用[5-6]。血管紧张素Ⅱ(angiotensin Ⅱ,AngⅡ)诱导心肌肥大是目前最常用的实验性心肌细胞肥大模型之一,其致心肌细胞肥大的机制尚未完全阐明。我们认为PERK介导的ERS可能是AngⅡ致心肌细胞肥大的机制。本工作在原代培养的乳大鼠心肌细胞AngⅡ诱导肥大模型上,研究ERS应激分子葡萄糖调节蛋白78 (glucose-regulated protein 78, GRP78)、钙网蛋白 (calreticulin, CRT)以及PERK途径相关分子表达的变化,证实PERK介导的ERS参与了AngⅡ诱导的心肌细胞肥大。
材 料 和 方 法
1动物与试剂
清洁级SD新生(出生24 h内)乳大鼠由军事医学科学院实验动物中心提供;DMEM培养基购自Gibco;新生牛血清购自PAA;胰蛋白酶购自Amresco;蛋白酶抑制剂购自Sigma;TRNzol-A+总RNA提取试剂、 2×Taq PCR MasterMix、电泳级琼脂糖,购自北京天根生化科技有限公司;cDNA第1链合成试剂盒,购自北京全式金生物技术有限公司;PCR引物由北京三博远志生物技术公司合成;TaqMan Gene Expression Master Mix和TaqMan荧光探针购自Applied Biosystems;蛋白电泳分子量(7~175 kD)标记为Bio-Rad产品;兔抗人CRT、 GRP78多克隆抗体、兔抗人GAPDH单克隆抗体和小鼠抗人CHOP单克隆抗体,购自Santa Cruz;兔抗人PERK和eIF2α多克隆抗体,购自Cell Signal;辣根过氧化酶标记山羊抗兔和山羊抗小鼠IgG购自Santa Cruz。
2乳鼠心肌细胞培养
按本室报道的方法[7]培养乳鼠心细胞:出生24 h内乳大鼠常规消毒后无菌操作取心尖部组织,剪碎成1 mm×1 mm×1 mm大小,加入适量0.15%胰蛋白酶,37 ℃水浴下机械振荡、反复消化,制备细胞悬液,差速贴壁法去除非心肌细胞,用含10%新生牛血清的DMEM培养液调整细胞浓度为3×109/L后接种于75 cm2培养瓶,置37 ℃、5% CO2孵箱进行原代培养。
在倒置相差显微镜下观察正常培养的SD乳大鼠心肌细胞,多呈不规则的多角形,少数有三角形及梭形。生长状态良好的心肌细胞在培养介质上贴壁较好,边缘折光率较低。以细胞免疫荧光检测生长于盖玻片的乳大鼠心肌细胞中的特异性标记物α-actin,同时以Hoechst 33258衬染细胞核,计数α-actin阳性细胞占总体细胞的百分数即为心肌细胞纯度(90%以上),见图1。

Figure 1. Morphological observation and identification of neonatal rat cardiomyocytes.A:under laser scanning confocal microcope;B:α-actin immunofluorescence assay and Hoechst 33258 staining.
图1乳鼠心肌细胞的形态观察和鉴定
3实验分组
取原代培养的心肌细胞,实验前24 h更换无血清DMEM培养基,随机分为以下7组:(1)正常对照(control)组:细胞常规培养至实验结束;(2)Ang Ⅱ组:培养液内加Ang Ⅱ(10-7mmol/L),常规培养48 h结束实验;(3)TG组:培养液内加入TG(50 mmol/L),常规培养48 h结束实验;(4)TM组:培养液内加入TM(10 μg/L),常规培养72 h结束实验;(5)Ang Ⅱ+Tau组:培养基中加入Tau(40 mmol/L),余同(2)组;(6)TG+Tau组:培养基中加入Tau(40 mmol/L),余同(3)组;(7)TM+Tau组:培养基中加入Tau(40 mmol/L),余同(4)组。
4蛋白质合成速率检测
采用[3H] -亮氨酸掺入技术测定心肌细胞蛋白质合成速率。实验结束前24 h,加入3.7×107Bq/L[3H] -亮氨酸孵育24 h。实验结束后用预冷4 ℃ PBS冲洗细胞2次,吸干废液,每孔内加入250 μL 甲酸,室温消化30 min后吹匀,全部移入液闪瓶。多功能液闪仪(Packard TRI-CARB 2100TR Liquid Scintillation Analyzer)检测心肌细胞[3H]-亮氨酸放射性强度,以每分钟校正计数(corrected counts per minute,ccpm)表示,单位为min-1。同时按照前述蛋白定量方法测定每组心肌细胞蛋白含量,以每毫克蛋白的ccpm反映各组蛋白质合成速率变化。
5心肌细胞表面积测定
细胞以1×104/cm2密度接种于24孔板内,以获得单个生长培养细胞,按照实验方法处理细胞后置于Olympus倒置显微镜下采集心肌细胞图像,每组中至少采集50个细胞,采用Image-Pro Plus 软件分析各组心肌细胞表面积。
6RT-PCR和实时定量PCR测定
采用TRNzol-A+提取总RNA,紫外分光光度计测定RNA含量,琼脂糖电泳显示清晰的18S和28S RNA荧光条带。按cDNA第1链合成试剂盒操作步骤将RNA反转录成cDNA,进行PCR扩增。各目的基因上、下游引物(P1、P2)见表1。电泳图像采用Image-Pro Plus 4.1图像分析软件分析平均吸光度值,以目的片段和GAPDH的平均吸光度比值反映目的基因的mRNA相对水平。以GAPDH为内参照,利用Taqman荧光探针和荧光定量PCR仪(ABI 7500 Fast Real-time PCR System),采用2-ΔΔCt相对定量法精确定量各目的基因的表达。
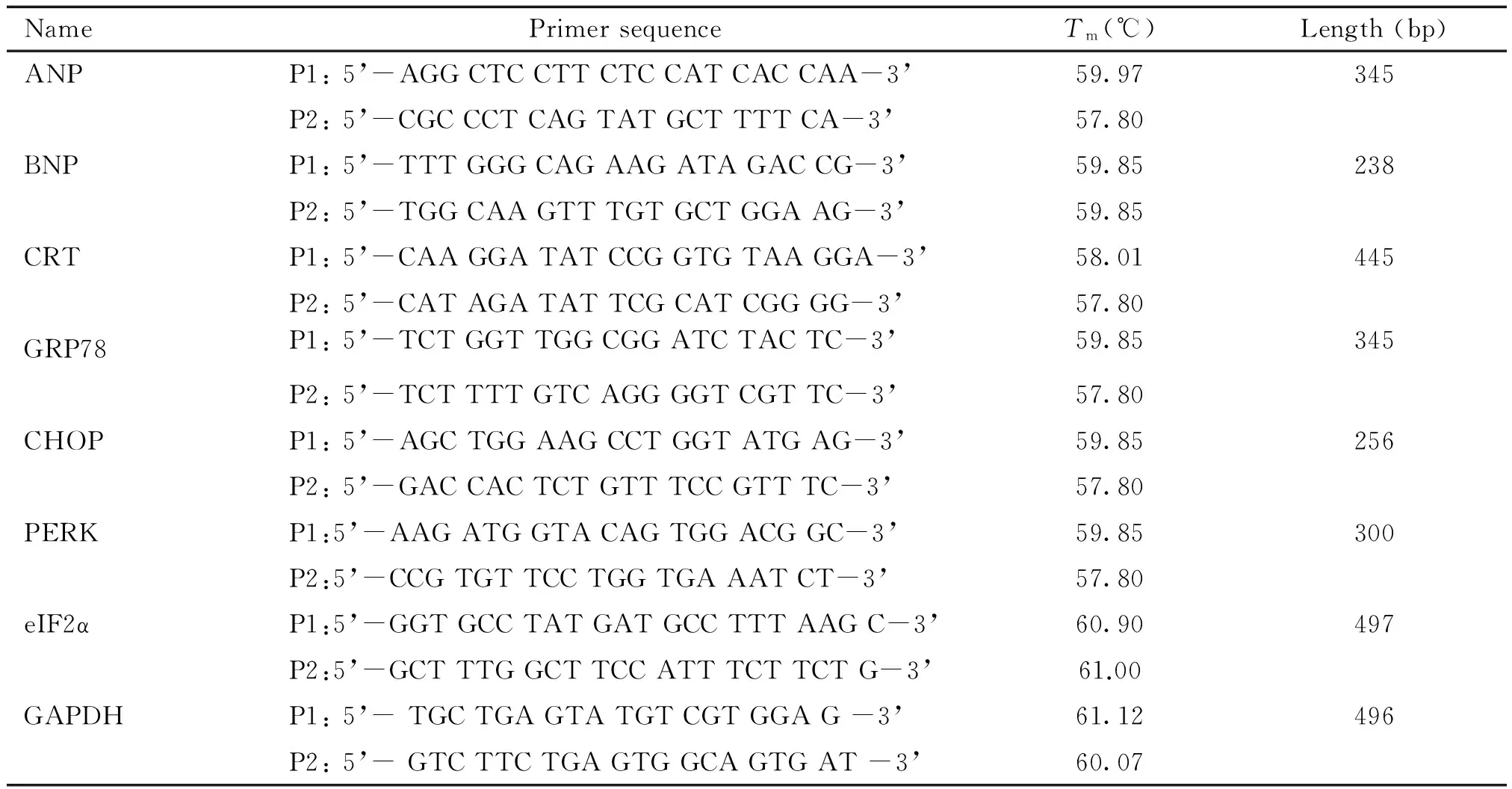
表1 PCR反应中目的片段引物
7Westernblotting检测
按本室报道方法[8]提取心肌细胞总蛋白,Bradford法蛋白定量后分装,-80℃保存。取上述细胞蛋白提取液上清 (含蛋白80 μg) 进行聚丙烯酰胺凝胶电泳 (SDS-PAGE,10%分离胶),将电泳分离后的蛋白质电转移至硝酸纤维素膜上,用5%BSA封闭40 min后分别加入CRT、GRP78、PERK、eIF2α多克隆抗体(均为1∶500)、CHOP单克隆抗体 (1∶500) 4 ℃过夜孵育,用1×TBS-T洗膜后,以相应的Ⅱ抗孵育1 h,并以GAPDH (1∶500) 单克隆抗体重复上述实验,作为上样对照。抗原-抗体复合物用ECL法显示,暗室X光胶片曝光,采用Image-Pro Plus 图像软件分析蛋白条带的积分吸光度值 (integrated absorbance,IA=平均吸光度值×面积),以靶蛋白IA值/GAPDHIA值的比值反映靶蛋白相对水平。
8统计学处理

结 果
1AngⅡ和ER应激诱导剂诱导心肌细胞显著肥大
1.1心肌细胞ANP和BNP mRNA表达的变化 采用RT-PCR检测心肌细胞肥大标志蛋白ANP和BNP mRNA表达改变如图2。结果显示10-7mmol/L AngⅡ、50 mmol/L TG分别作用48 h以及10 μg/L TM作用72 h后心肌细胞ANP和BNP mRNA表达均显著升高。其中10-7mmol/L AngⅡ组ANP和BNP mRNA表达分别较对照组高147.0%和154.5%(P<0.05),50 mmol/L TG组分别较对照组高210.1%和142.9%(P<0.05),10 μg/L TM组分别较对照组高189.0%和284.8%(P<0.05)。内质网应激抑制剂牛磺酸明显抑制AngⅡ和内质网应激诱导剂TG、TM诱导的心肌细胞ANP和BNP表达上调,其中AngⅡ+Tau组较AngⅡ组分别低57.5%和38.4%(P<0.05),TG+Tau组较TG组分低57.1%和31.3%(P<0.05),TM+Tau组较TM组分别低45.3%和68.3%(P<0.05)。


图2AngⅡ、ERS诱导剂以及加入牛磺酸后对ANP和BNPmRNA表达的影响
1.2心肌细胞蛋白质合成速率的变化 [3H]-亮氨酸掺入技术检测心肌细胞蛋白质合成速率变化结果见图3。与对照组比较,10-7mmol/L AngⅡ、50 mmol/L TG分别作用48 h以及10 μg/L TM作用72 h后心肌细胞蛋白质合成速率分别较对照组高134.8%、156.6%和136.1%(P<0.05)。内质网应激抑制剂牛磺酸明显抑制AngⅡ和内质网应激诱导剂TG、TM诱导的心肌细胞蛋白质合成增加,其中AngⅡ+Tau组较AngⅡ组蛋白质合成速率低32.5%(P<0.05),TG+Tau组较TG组蛋白质合成速率低33.4%(P<0.05),TM+Tau组较TM组蛋白质合成速率低31.1%(P<0.05)。

图3AngⅡ、ERS诱导剂以及加入牛磺酸后对蛋白质合成速率的影响
1.3心肌细胞表面积的变化 心肌细胞表面积测定结果见图4。正常对照组细胞生长状态良好,10-7mmol/L AngⅡ、50 mmol/L TG分别作用48 h以及10 μg/L TM作用72h后心肌细胞表面积分别较对照组高99.7%、78.2%和88.0%(P<0.05)。内质网应激抑制剂牛磺酸明显抑制AngⅡ和内质网应激诱导剂TG、TM诱导的心肌细胞表面积增加,其中AngⅡ+Tau组较AngⅡ组表面积低33.0%(P<0.05),TG+Tau组较TG组蛋白表面积低26.5%(P<0.05),TM+Tau组较TM组表面积低38.2%(P<0.05)。
2AngⅡ对心肌细胞ERS反应的影响
2.1CRT、GRP78和CHOP表达 采用实时定量PCR和Western blotting检测内质网应激分子CRT、GRP78和CHOP mRNA和蛋白表达结果见图5。与对照组比较,10-7mmol/L AngⅡ、50 mmol/L TG分别作用48 h以及10 μg/L TM作用72 h后CRT mRNA表达分别高146.4%、290.5%和184.5%(P<0.05),蛋白表达分别高125.3%、158.6%和105.1%(P<0.05)。上述3组的GRP78 mRNA表达分别高84.0%、70.8%和98.2%(P<0.05),蛋白表达分别高77.6%、48.1%和74.2%(P<0.05)。上述3组的CHOP mRNA表达分别高117.7%、142.2%和210.4%(P<0.05),蛋白表达分别高63.3%、117.2%和122.5%(P<0.05)。内质网应激抑制剂牛磺酸明显抑制AngⅡ和内质网应激诱导剂TG、TM诱导的心肌细胞内质网应激反应,与AngⅡ组比较, AngⅡ+Tau组CRT、GRP78和CHOP mRNA表达分别低57.6%、60.6%和62.7%(P<0.05),蛋白表达分别低43.1%、33.3%和37.4%(P<0.05)。TG+Tau组CRT、GRP78和CHOP mRNA表达分别较TG组低57.1%、75.9%和61.2%(P<0.05),蛋白表达分别低54.3%、30.4%和38.2%(P<0.05)。TM+Tau组CRT、GRP78和CHOP mRNA表达分别较TM组低54.5%、70.4%和74.8%(P<0.05),蛋白表达分别低30.2%、38.0%和41.0%(P<0.05)。

图4AngⅡ、ERS诱导剂以及加入牛磺酸后对心肌细胞表面积的影响
2.2PERK和eIF2α表达 采用RT-PCR和Western blotting检测PERK和eIF2α mRNA和蛋白质表达结果见图6。与对照组比较,10-7mmol/L AngⅡ、50 mmol/L TG分别作用48 h以及10 μg/L TM作用72 h后PERK mRNA表达分别高165.4%、131.9%、209.2%(P<0.05),蛋白表达分别高132.1%、117.3%、205.1%,P<0.05。上述3组的eIF2α mRNA表达分别较对照组高110.9%、169.6%、112.0%,P<0.05,蛋白表达分别高46.5%、82.8%、73.2%,P<0.05。内质网应激抑制剂牛磺酸明显抑制AngⅡ和内质网应激诱导剂TG、TM诱导的心肌细胞PERK和eIF2α表达上调,与AngⅡ组比较,AngⅡ+Tau组PERK和eIF2α mRNA表达分别低31.7%和56.2%(P<0.05),蛋白表达分别低43.5%和17.8%(P<0.05)。与TG组比较,TG+Tau组PERK和eIF2α mRNA表达分别低40.0%和61.0%(P<0.05),蛋白表达分别低35.9%和45.1%(P<0.05)。与TM组比较,TM+Tau组PERK和eIF2α mRNA表达分别低38.8%和56.6%(P<0.05),蛋白表达分别低44.9%和24.9%(P<0.05)。

图5AngⅡ、ERS诱导剂以及加入牛磺酸后对CRT、GPR78和CHOP表达的影响

图6AngⅡ、ERS诱导剂以及加入牛磺酸后对PERK和eIF2α表达的影响
讨 论
心肌肥大的主要表现为细胞体积增大、重量及蛋白质合成速率增加以及间质细胞增生[9],并出现ANP和BNP表达的动态变化,其中ANP被认为是心肌肥大的标志分子[10],而BNP可以预测心力衰竭发生,被认为是心力衰竭的标志分子[11]。本工作在培养的乳大鼠心肌细胞AngⅡ诱导心肌肥大模型上,证实AngⅡ(10-7mmol/L作用48 h)使心肌细胞表面积、蛋白质合成速率及ANP和BNP表达均上调,表明AngⅡ明显诱导心肌细胞肥大,与文献报道一致[12]。
AngⅡ致心肌肥大的机制尚未完全阐明,近年来内质网应激在心肌肥大发病机制中的作用开始受到重视[13]。课题组前期研究结果显示,ERS诱导剂TG和TM呈时间和剂量依赖性诱导心肌细胞发生ERS和心肌细胞肥大,证实ERS是诱导心肌细胞肥大的重要因素[4]。ERS是一种在进化上高度保守的细胞反应机制。适度的ERS有利于增强ER处理未折叠蛋白或错误折叠蛋白的能力,促进钙稳态的恢复:持续而严重的ERS可明显上调GPR78和CRT表达,诱导激活PERK介导的ERS相关凋亡途径[14-16]。本研究发现,以10-7mmol/L AngⅡ处理心肌细胞48 h,GRP78和CRT的mRNA和蛋白表达明显升高,与50 mmol/L TG和10 μg/L TM分别作用48 h和72 h后GRP78和CRT的变化相似,与文献报道相一致[17]。提示AngⅡ诱导心肌细胞肥大过程中发生严重ERS。内质网应激抑制剂牛磺酸明显抑制AngⅡ诱导的心肌细胞内质网应激反应,表现为GRP78和CRT mRNA和蛋白表达显著降低,与文献报道相一致[18],并消除AngⅡ诱导心肌细胞肥大。内质网应激抑制剂牛磺酸还抑制TG和TM诱导的心肌肥大,表现为ANP和BNP表达、心肌细胞表面积减少以及蛋白质合成速率降低,上述结果提示ERS参与了AngⅡ诱导的心肌细胞肥大。
PERK是介导内质网应激反应的重要感受分子,正常状态下与GRP78结合成无活性的复合物; ERS时GRP78与未折叠蛋白或错误折叠蛋白结合后,解离出PERK,使其寡聚化并激活,进而磷酸化eIF2α,下调mRNA和蛋白的转录翻译水平,减轻ER蛋白负荷[19]。持续激活PERK/eIF2α信号通路将上调CHOP的转录翻译水平,最终引起细胞凋亡[20-21]。本研究发现,以10-7mmol/L AngⅡ处理心肌细胞48 h,PERK、eIF2α和CHOP的mRNA和蛋白表达明显升高,与50 mmol/L TG和10 μg/L TM分别作用48 h和72 h后PERK、eIF2α和CHOP的变化相一致,提示ERS中的PERK/eIF2α信号途径参与了AngⅡ诱导心肌细胞的肥大过程,同时持续的ERS可能启动了CHOP介导的细胞凋亡途径[22]。培养液中加入牛磺酸后,发现各组中的PERK、eIF2α和CHOP mRNA和蛋白表达显著降低,提示牛磺酸可能通过调控PERK/eIF2α信号途径的上游靶点抑制PERK激活,从而减轻ERS。上述结果表明PERK介导的ERS参与了AngⅡ诱导的心肌细胞肥大。
[1] Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus[J]. Physiol Rev, 1999,79(3):683-701.
[2] DeGracia DJ, Montie HL. Cerbral ischemia and the unfolded protein response[J]. J Neurochem, 2004,91(1):1-8.
[3] 李载权,周爱儒,唐朝枢. 内质网应激反应分子机理研究进展[J]. 中国生物化学与分子生物学报, 2005,20(3):283-288.
[4] Zhang ZY, Liu XH, Hu WC, et al. The calcineurin-myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes[J]. Am J Physiol Heart Circ Physiol. 2010,298(5):H1499-H1509.
[5] Guz G, Oz E, Lorttar N, et al. The effect of taurine on renal ischemia-reperfusion injury[J]. Amino Acids, 2007,32(3):405-411.
[6] Nonaka H, Tsujino T, Watari Y, et al. Taurine prevents the decrease in expression and secretion of extracellular superoxide dismutase induced by homocysteine: amelioration of homocysteine-induced endoplasmic reticulum stress by taurine[J]. Circulation, 2001,104(10):1165-1170.
[7] Liu XH, Wu XD, Cai LR, et al. Hypoxic preconditioning of cardiomyocytes and cardioprotection: phosphorylation of HIF-1α induced by p42/p44 mitogen-activated protein kinases is involved [J]. Pathophysiology, 2003, 9(4): 201-205.
[8] Liu XH, Wu XD, Han Y, et a1. Signal pathway of cardioprotection induced by monophosphoryl lipid A in rabbit myocardium [J]. Pathophysiology,2002,8(3): 193-196.
[9] Krauser DG, Devereux RB. Ventricular hypertrophy and hypertension: prognostic elements and implications for management[J]. Herz,2006,31(4): 305-316.
[10]Melo LG, Pang SC, Ackermann U. Atrial natriuretic peptide: regulator of chronic arterial blood pressure[J].News Physiol Sci,2000,15:143-149.
[11]Tang WH, Francis GS, Morrow DA, et al. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure[J]. Circulation,2007,116(5):e99-e109.
[12]徐菲菲,刘秀华,王彦珍. 肌原纤维调节因子-1在心肌肥大中的作用研究[J]. 中国病理生理杂志,2006,22(3):443-447.
[13]徐占稳,张苏河. 血管紧张素与心肌肥大的相关性研究进展[J]. 实用医学杂志,2008,24(11):2010-2011.
[14]Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticolum: coordination of gene transcriptional and translational controls [J]. Gene Dev, 1999,13(10): 1211-1233.
[15]Liu XH. Progress in endogenous cardioprotection induced by ischemia postconditioning[J]. Acta Physiol Sin, 2008,59(5):628-634.
[16]刘秀华. 心力衰竭的内质网应激机制[J]. 中国病理生理杂志, 2010,26(10):1964.
[17]Okada K, Minamino T, Minamino Y, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction:possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis[J]. Circulation,2004,110(6):705-712.
[18]Song H, Kim H, Park T, et al. Characterization of myogenic differentiation under endoplasmic reticulum stress and taurine treatment[J]. Adv Exp Med Biol,2009,643:253-261.
[19]Liu CY, Schroder M, Kaufman RJ. Ligand-independent dimmerization activates the stress-response kinase IRE1 and PERK in the lumen of the endoplasmic reticulum[J]. J Biol Chem, 2000,275(32):24881-24885.
[20]Lin JH, Li H, Zhang Y, et al. Divergent effects of PERK and IRE1 signaling on cell viability[J]. PLoS One, 2009,4(1):e4170.
[21]Breckenridge DG, Germain M, Mathai JP, et al. Regulation of apoptosis by endoplasmic reticulum pathways [J]. Oncogene, 2003,22(53): 8608-8618.
[22]Brostrom MA,Mourad F,Brostrom CO,et al. Regulated expression of GRP78 during vasopressin-induced hypertrophy of heart-derived myocytes[J].J Cell Biochem,2001,83(2):204-217.
PERK-mediatedendoplasmicreticulumstressisinvolvedinangiotensinII-inducedmyocardialhypertrophy
LÜ Zhen-rong1, WANG Xiao-reng2, LI Yu-zhen2, WANG Chen2, LIU Xiu-hua2
(1DepartmentofPathophysiology,MedicalSchoolofShandongUniversity,Jinan250012,China;2DepartmentofPathophysiology,ChinesePLAGeneralHospital,Beijing100853,China.E-mail:xiuhualiu98@yahoo.com.cn)
AIM: To investigate the role of protein kinase R-like endoplasmic reticulum kinase (PERK)-mediated endoplasmic reticulum stress (ERS) in angiotensin II (AngⅡ) -induced myocardial hypertrophy.METHODSIn the hypertrophy model of AngⅡ-induced cardiomyocytes isolated from neonatal Sprague-Dawley rats, the methods of morphological observation, [3H]-leucine incorporation and surface area measurement were employed to assess the cardiomyocyte hypertrophy. Real-time PCR, RT-PCR and Western blotting were used to detected the expression of glucose-regulated protein 78 (GRP78), calreticulin (CRT), PERK, eukaryotic initiation factor 2α (eIF2α) and C/EBP homologous protein (CHOP) at mRNA and protein levels.RESULTSCompared with control group, Ang II-treated cardiomyocytes showed that the mRNA and protein expression of CRT increased by 146.4% and 125.3%, respectively (P<0.05). The mRNA and protein expression of GRP78 increased by 84.0% and 77.6%, respectively (P<0.05). The mRNA and protein expression of PERK increased by 165.4% and 132.1%, respectively (P<0.05).The mRNA and protein expression of eIF2α was increased by 110.9% and 46.5%, respectively (P<0.05). The mRNA and protein expression of CHOP also increased by 117.7% and 63.3%, respectively (P<0.05).CONCLUSIONPERK-mediated ERS response is involved in AngⅡ-induced cardiomyocyte hypertrophy.
Cardiac hypertrophy; Endoplasmic reticulum stress; Protein kinase R-like endoplasmic reticulum kinase
R363.1
A
10.3969/j.issn.1000-4718.2012.07.001
猜你喜欢
国外畜牧学(猪与禽)(2022年1期)2022-07-10
电脑报(2022年25期)2022-07-05
江苏农业科学(2022年2期)2022-02-15
饲料工业(2021年6期)2021-04-16
江西农业学报(2021年1期)2021-02-01
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
北华大学学报(自然科学版)(2020年5期)2020-10-21
食品与健康(2020年7期)2020-07-09
上海医药(2016年23期)2016-12-22
