
慢病毒介导的猪CD4启动子在白血病 Jurkat 细胞中的启动作用
2012-10-10 09:24林晓晨曹宇航任林柱逄大欣
吉林大学学报(医学版) 2012年2期
林晓晨,杨 鑫,曹宇航,焦 丽,戴 甄,任林柱,逄大欣
(吉林大学畜牧兽医学院 动物胚胎工程吉林省重点实验室,吉林 长春 130062)
CD4是一种跨膜糖蛋白,主要表达于辅助T细胞表面,是TCR识别抗原的供受体,与MHCⅡ类分子多肽区结合[1]。目前已克隆鉴定了人、猴、鼠、兔、牛、猫、狗等动物的CD4-DNA序列。20世纪80年代,应用单克隆抗体的免疫组织化学技术与流式细胞技术在猪的T淋巴细胞表面鉴定了CD4分子。CD4基因的表达受到多基因的控制[2],其中包括启动子、增强子及沉默子等调控元件[3]。为了能使CD4基因更好地表达,使其他物种启动子为人类疾病研究起到辅助作用,CD4基因调控原件的研究一直成为研究重点。CD4分子以单体的形式存在,CD4+T 细胞能识别MHCⅠ类分子递呈的抗原,它们除了作为细胞黏连分子发挥作用外,还是传递信号的分子,具有信号传递功能[4-5]。基因转录是遗传信息传递和表达的枢纽,是基因表达调控机制发挥作用的重要环节[1]。而启动子是决定转录起始点和转录频率的关键元件,因此启动子的识别对整个基因组功能的诠释具有重要作用[6]。启动子的研究对于阐释基因表达调控网络的机制和基因组的功能都具有非常重要的意义。启动子的结构影响了其与RNA聚合酶的亲和力,从而影响基因表达水平。转录单元(transcription unit) 是一段从启动子开始至终止子(terminator)结束的DNA序列,RNA聚合酶从转录起点开始沿着模板前进,直到终止子为止,转录出一条RNA链。在细胞中,一个转录单元可以是一个基因,也可以是几个基因。启动子区是RNA聚合酶的结合区,其结构直接关系到转录的效率[7-8]。研究启动子,也就是要研究其在细胞中启动外源基因的能力,而对于较难转染的细胞来说,现在有—种新型的转染方式——慢病毒侵染。慢病毒载体是HIV-1源性的复制缺陷型逆转录病毒,采用表达VSV包膜蛋白取代了HIV本身包膜蛋白,使其大大提高了转导效率,并扩展了感染宿主细胞的范围,能感染所有组织来源的分裂和非分裂细胞,携带的目的基因能整合到宿主细胞基因组,使得外源基因获得长期稳定表达。目前,体内外实验中均未发现慢病毒载体对正常组织细胞有毒副作用。本实验通过慢病毒表达载体研究异种基因启动子在Jurkat细胞中调节外源基因表达的作用,该系统能在较长时间里明显地观察到外源基因的表达情况,本实验旨在为建立人类白血病动物模型奠定理论基础。
1 材料与方法
1.1 主要试剂
PGM-T载体购自Invitrogen公司;Tag酶 T4DNA聚合酶和T4DNA连接酶购自 TAKARA公司;质粒小抽试剂盒 胶回收试剂盒和无内毒素质粒大抽试剂盒购自TIANGEN公司;NotⅠ 和EcoRⅠ 购自TAKARA公司;慢病毒包装系统和293T包装细胞购自Cell biolabs公司;Jurkat细胞由吉林大学白求恩医学院免疫学实验室提供;RPMI 1640、DMEM 培养基、胎牛血清、谷氨酰胺、 非必需氨基酸、青霉素、链霉素和胰蛋白酶均购自美国Invitrogen公司;质粒小抽试剂盒、胶回收试剂盒和无内毒素质粒大抽试剂盒均购自 TIANGEN公司;Lipofectamine 2000购自Invitrogen公司;引物由北京TIANGEN公司合成。
1.2 引物设计合成
根据查找的猪CD4基因序列进行设计,SALMON等[8]已经得出人和大鼠CD4基因启动子具有相同的5对保守序列。首先查找猪CD4全序列,通过5对保守序列与猪CD4基因前1 400 bp比较,发现猪基因组中也同样包含有与人、鼠相同的保守序列。另外,由于家猪与野猪在CD4基因启动子与增强子序列上存在差别,所以通过启动子在线分析软件和从文献中得到的序列,获得家猪可能的启动子与增强子范围,利用引物设计软件Primer 5.0设计引物,上游:5′-GCGGCCGCGATCTCCAGAGATCAAACC-3′;下游:5′-GAATTCAAGCGAGTCTCTGCAGAAGTA -3′,在上、下游引物加入NotⅠ和EcoRⅠ 2个酶切位点(下划线部分),并由北京TIANGEN公司合成。
1.3 基因的克隆及载体的构建
提取长白猪的血液基因组作为PCR模板,利用6对引物进行PCR,反应体系为E*Tag0.5 μL,模板1 μL,引物各1 μL,DNTP 4 μL,Buffer 5 μL,ddH2O 37.5 μL,共50 μL。得到的产物回收并连接到PGM-T载体上进行测序。将测序结果与查找结果进行比对,并与猪全基因组进行比对,分析该片段是否正确。将慢病毒包装系统主质粒PSMPUW的启动子EF-1α利用NotⅠ和EcoRⅠ 2个酶切位点切掉,连入已经克隆得到的猪CD4基因启动子,得到载体PSMPUW-SP,并比对确认连入的片段是否出现偏差。用此载体进行慢病毒包装级检验实验。
1.4 细胞培养
Jurkat细胞株,于37℃ 、5% CO2条件下培养。培养液为含有10%FBS、1%双抗、1%谷氨酰胺替代物和1%非必须氨基酸的RPMI 1640培养基。Jurkat细胞为悬浮培养的癌细胞,生长速度很快,1~2 d就必须传代1次。否则其培养基很难满足其消耗致使细胞可能出现大量死亡或者生长状态缓慢。293T细胞株,半悬浮半贴壁细胞,于37℃、5% CO2条件下培养。培养基为包含有10%FBS,1%双抗,1%谷氨酰胺替代物,1%非必须氨基酸的DMEM培养基。
1.5 慢病毒包装、测定及侵染
1.5.1 慢病毒的包装 提前1 d将293T细胞接种到6孔板,当细胞达到70%~80%汇合时进行实验。利用DPBS清洗选定的转染孔2次,重新加入新鲜培养基待用。将慢病毒主质粒和辅助质粒以3∶1∶1∶1比例与相应体积的Opti-MEM混合均匀,与稀释后的Lipofectamine 2000进行混合,形成DNA与Lipofectamine 2000的转染复合物。将Lipofectamine 2000-DNA转染复合物加入到准备好的293T细胞中,于37℃、5% CO2条件下培养6~8 h后换入带有10%FBS的培养液。24和48 h后绿色荧光显微镜下检测转染效率,于37 ℃、5% CO2培养箱内继续培养48 h。收集转染后48 h的293T细胞上清液,于4℃、4 000 r·min-1离心10 min,除去细胞碎片。以0.45 μm皿滤器过滤上清液于15 mL离心管中。放于-70℃保存待用。
1.5.2 慢病毒滴度的测定和侵染 在青霉素瓶或离心管中将保存的含有病毒的上清液作连续5倍的稀释,从10-1到10-5。将稀释好的病毒接种到96孔微量培养板中,每一稀释度接种一纵排共8 孔,每孔加入细胞悬液100 μL,使细胞量达到(2~3)×105mL-1。设正常细胞对照,正常细胞对照作两纵排(100 μL生长液+100 μL细胞悬液)。逐日观察并记录出现阳性结果的孔数,一般需要观察5~7 d。根据下列公式计算半数组织培养感染剂量(TCID50)。Reed-Muench两氏法:距离比例=(高于50%的病变率的百分数-50%)/(高于50%的病变率的百分数-低于50%的病变率的百分数);lgTCID50=距离比例×稀释倍数之间的差+高于50%的病变率的稀释对数。Karber法:lgTCID50=最高稀释度的对数-稀释度对数之间的差×(阳性孔比率总和-0.5)。
将Jurkat细胞计数,选取(1~2)×105的细胞换液后加入到24孔板中,20~24 h选取细胞最佳生长状态时,将细胞培养液完全换成包涵病毒的培养液。左右轻摇细胞使细胞达到均匀分布为最佳,置于37℃ 、5% CO2条件下培养24~48 h后绿色荧光显微镜下观察结果。
2 结 果
2.1 猪CD4启动子的克隆及慢病毒载体的构建
利用启动子在线分析软件查找前1 400 bp片段序列可能出现启动子的位置,见图1。由此设计引物,以血液提取出的基因组作为模板进行PCR反应,得到猪CD4启动子序列,并且将PCR产物连接T载体,得到PGM-T-SP,进行测序并鉴定,鉴定结果见图2。由于家猪与野猪基因不同,得到的结果与查找得到野猪的基因相似度在50%以上,得到的PCR产物包含5对保守序列。将得到的猪CD4基因启动子片段利用NotⅠ和EcoRⅠ 2个酶切位点与利用2个酶切位点切掉启动子的慢病毒主质粒进行连接,得到PSMPUW-SP,将其转化至TOP10感受态后经KAN抗性筛选阳性质粒,提取质粒后进行酶切鉴定,鉴定结果见图3。慢病毒表达载体的构建结果见图4。
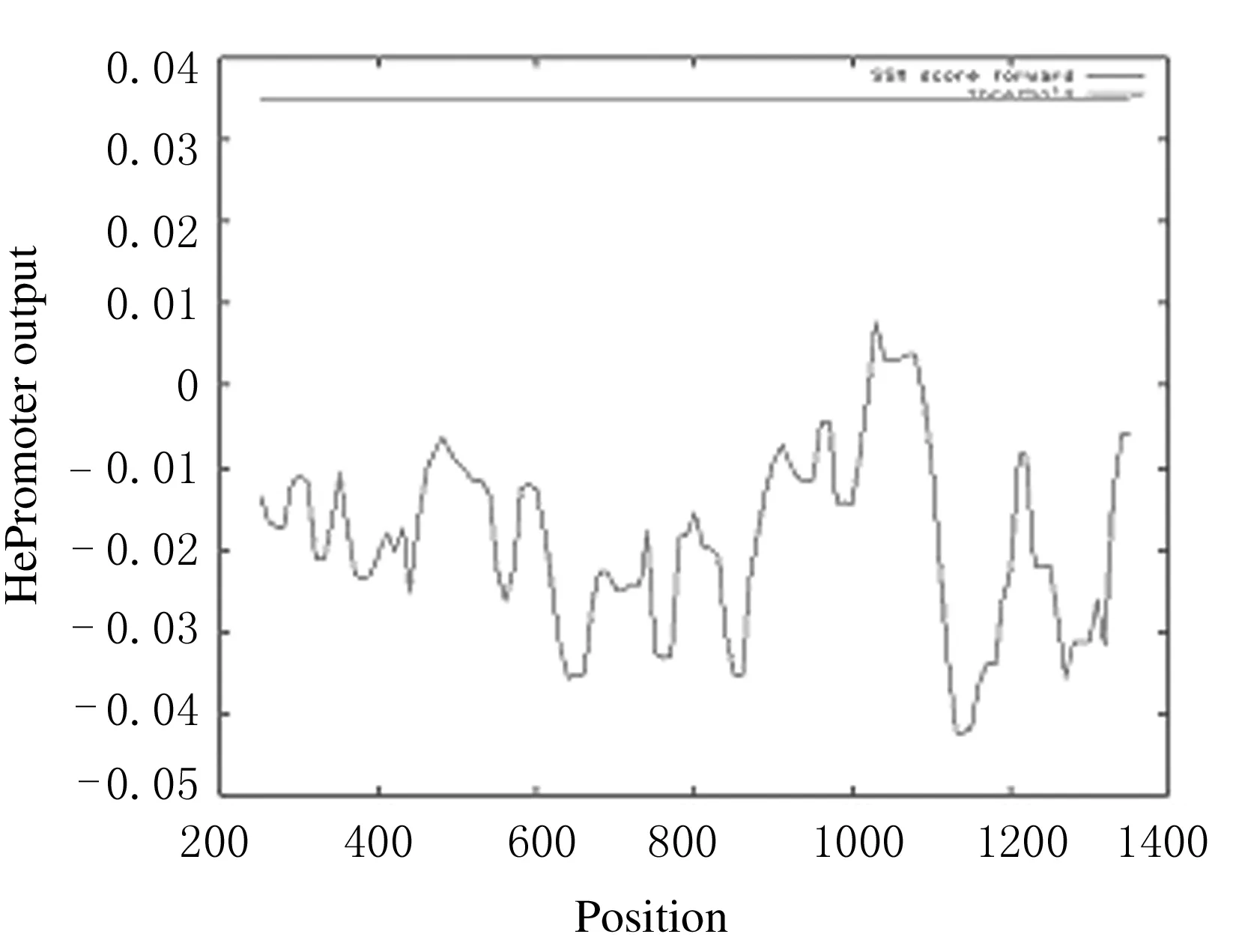
图1 全基因前1 400 bp序列预测出的启动子位置

图2 SP的PCR结果(A) 和PGM-T-SP的鉴定结果(B)
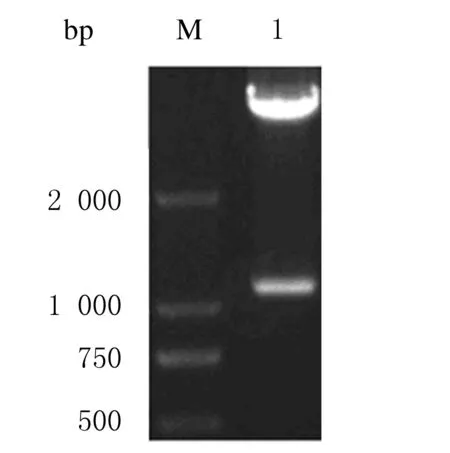
图3 PSMPUW-SP的酶切鉴定结果
2.2 慢病毒包装结果
培养的293T细胞包装病毒后,利用绿色荧光显微镜进行观察,携带CD4基因启动子的慢病毒包装体系中的主质粒进入到293T细胞中,绿色荧光表达量很明显。相对于EF-1α启动子启动的原质粒,猪CD4基因启动子在293T细胞中能更好地表达。CD4基因启动子与EF-1α启动子在293T细胞中的启动效果没有很大差别,说明猪CD4基因启动子启动的高效性。见图5(封二)。
2.3 慢病毒滴度测定
结果显示:病毒稀释液浓度为10-1时出现8个阳性孔数,10-2时出现8个阳性孔数,10-3时出现6个阳性孔数,10-4时出现3个阳性孔数,10-5时出现0个阳性孔数;通过累加,病毒稀释液浓度为10-1~10-5时分别出现25、17、11、3和0个阳性孔数。由此计算阳性孔数占总孔数的比例,10-1~10-5分别为100.0%、100.0%、84.6%、30.0%和0%。根据公式计算,Reed-Muench两氏法:距离比例(84.6-50.0)/(84.6-30.0)=0.6,lgTCID50=0.6×(-1)+(-3)=-3.6,即TCID50=10-3.6·0.1 mL-1。Karber法:lgTCID50=-1-1×(3.125-0.5)=-3.625,同样可以说明TCID50=10-3.6·0.1 mL-1。综合2种计算结果得出大概的病毒量为10-3.6·0.1 mL-1。
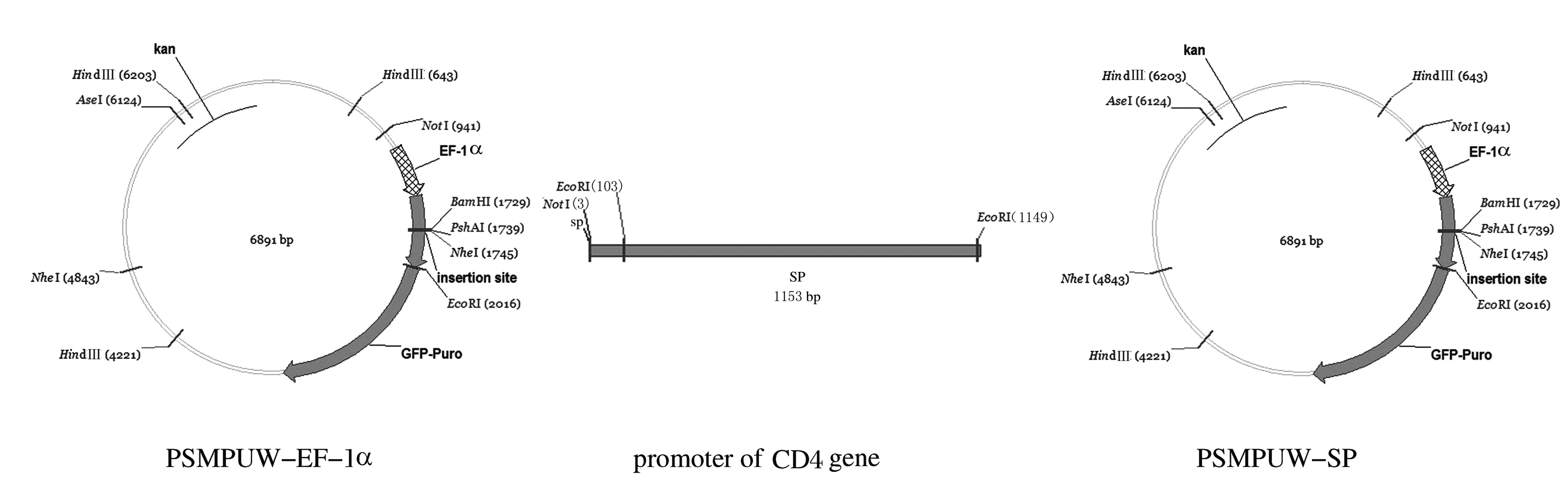
图4 慢病毒表达载体构建示意图
2.4 侵染结果照相
利用包装出来的病毒侵染了2种细胞,侵染的293T细胞中可见成功包装的慢病毒,见图6(封二)。虽然包装出来的病毒并没有达到理想值,但其感染效果很明显,绿色荧光表达很好,而且慢病毒表达时间较长的特性也能利于更好地观测和得出结论。从侵染Jurkat细胞后对绿色荧光蛋白的观察可见:CD4基因启动子包装出来的病毒与EF-1α启动子包装出来的病毒的阳性病毒相近,见图7(封二)。结果显示:克隆得到的猪CD4基因启动子也许并非是一种特异性启动子,其与人、大鼠CD4基因启动子从遗传特性上有着很大的不同。2种细胞侵染结果对比显示:Jurkat细胞具有难转染的特性,普通的转染试剂很难达到效果。
3 讨 论
猪作为一种模型生物有着很多优点,并且越来越受到研究人员的关注,其生理特征和病理特征均可以作为研究的对象。本实验对猪CD4基因启动子进行预测及克隆,并采用慢病毒表达载体对克隆得到的启动子进行验证[9]。结果显示其在293T细胞中可以大量表达,而且在Jurkat细胞中得到了同样的结论。虽然人和大鼠的CD4基因启动子在淋巴细胞中能够特异表达,但研究发现:猪CD4基因启动子也许并非是一种特异性启动子。目前,区别特异性启动子与非特异性启动子的方法,均是以基因在某种特定细胞中的表达量的多少来区分。以CMV启动子为例,CMV启动子来源于病毒,相对与内源性的启动子,在哺乳动物细胞中CMV启动子表达可能更易于受到细胞内沉默机制的影响[10]。CMV启动子序列中含有NFκB 结合位点,NF 位点可以明显增强CMV 启动子启动基因序列的表达水平[11],然而CMV启动子启动外源基因表达并非是最好的选择,真核基因的表达调控是一个复杂的动态过程,在转录起始、转录延伸和转录终止的循环中,每一个细节都受到精细而复杂的调控。基因启动子在转录起始过程中发挥重要的调控作用,其能够被很多反式作用因子识别,在不同细胞中,同一个启动子因相互作用的反式作用因子不同,基因的表达水平也不同。而在同一细胞中,相同反式作用因子对不同启动子的识别序列不一致。在某些细胞中这些组成型启动子驱动的基因表达强度存在着差异。在小鼠胚胎干细胞中 CMV启动子驱动基因的表达能力很弱,而伴随着细胞的分化,CMV驱动基因的表达能力明显增加。因此在目的细胞中筛选最适启动子非常必要,转录起始的能力也并不相同。虽然在很多细胞中CMV启动基因的表达效果最强,然而许多实验[12-14]结果表明:CMV启动子并不能在机体组织中长期持续表达。
如果说CMV并非特异性,那么是否也可以将在人癌细胞中同样高表达的猪CD4基因启动子作为非特异性启动子?本实验结果显示:猪CD4基因启动子和EF-1α启动子启动绿色荧光蛋白在细胞中表达量虽然有所不同,但总体趋势不变。SP与EF-1α在相同细胞当中有着同样的表达效果。猪CD4基因启动子与已经确定的人、鼠CD4基因特异性启动子有区别,空间结构的不确定,反式作用因子的识别等都是制约启动子在细胞中发挥作用的因素。实验结果显示真核细胞基因启动子在细胞中的转录的空间结构和转录后修饰状态是一定的,不同物种之间启动子在细胞中的表观遗传学修饰状态可能导致其驱动基因表达能力存在差异。本研究确定了猪CD4基因启动子的序列和其在293T和Jurkat细胞中启动绿色荧光蛋白基因的表达效果,在人类白血病癌细胞Jurkat中,CD4基因启动子启动效果与EF-1α启动子启动效果比较差别不大。可以得到这样的结论:猪CD4基因启动子并非具有特异性。
[参考文献]
[1]Itano A,Salmon P,Kioussis D,et al.The cytoplasmic domain of CD4 promotes the development of CD4 lineage T cells [J].J Exp Med,1996,183(3):731-741.
[2]Sarafova S,Siu G.Precise arrangement of factor-binding sites is required for murine CD4 promoter function [J].Nucleic Acids Res,2000,28(14):2664-2671.
[3]Wurster AL,Siu G,Leiden JM,et al.Elf-1 binds to a critical element in a second CD4 enhancer [J].Mol Cell Biol,1994,14(10):6452-6463.
[4]Chong MM,Simpson N,Ciofani M,et al.Epigenetic propagation of CD4 expression is established by the CD4 proximal enhancer in helper T cells [J].Genes Dev,2010,24(7):659-669.
[5]Duncan DD,Stupakoff A,Hedrick SM,et al.A Myc-associated zinc finger protein binding site is one of four important functional regions in the CD4 promoter [J].Mol Cell Biol,1995,15(6):3179-3186.
[6]Sarafova S,Siu G.A potential role for Elf-1 in CD4 promoter function [J].J Biol Chem,1999,274(23):16126-16134.
[7]Lo SF,Wan L,Lin HC,et al.Association of CD4 enhancer gene polymorphisms with rheumatoid arthritis and systemic lupus erythematosus in Taiwan [J].J Rheumatol,2008,35(11):2113-2118.
[8]Salmon P,Giovane A,Wasylyk B,et al.Characterization of the human CD4 gene promoter: transcription from the CD4 gene core promoter is tissue-specific and is activated by Ets proteins [J].Proc Natl Acad Sci USA,1993,90(16):7739-7743.
[9]李振宇,徐开林,潘秀英,等.慢病毒载体介导绿色荧光蛋白基因在小鼠T淋巴细胞中的表达 [J].中国实验血液学杂志,2007,15(1):125-128.
[10]Gill DR,Smyth SE,Goddard CA,et al.Increased persistence of lung gene expression using plasmids containing the ubiquitin C or elongation factor 1alpha promoter [J].Gene Ther,2001,8(20):1539-1546.
[11]Laegreid A,Medvedev A,Nonstad U,et al.Tumor necrosis factor receptor p75 mediates cell-specific activation of nuclear factor kappa B and induction of human cytomegalovirus enhancer [J].J Biol Chem,1994,269(10):7785-7791.
[12]Guo ZS,Wang LH,Eisensmith RC,et al.Evaluation of promoter strength for hepatic gene expression in vivo following adenovirus-mediated gene transfer [J].Gene Ther,1996,3(9):802-810.
[13]Loser P,Jennings GS,Strauss M,et al.Reactivation of the previously silenced cytomegalovirus major immediate-early promoter in the mouse liver: involvement of NFkappaB [J].J Virol,1998,72(1):180-190.
[14]Yew NS,Wysokenski DM,Wang KX,et al.Optimization of plasmid vectors for high-level expression in lung epithelial cells [J].Hum Gene Ther,1997,8(5):575-584.
猜你喜欢
当代水产(2022年1期)2022-04-26
农业科技通讯(2021年3期)2021-04-04
载人航天(2020年5期)2020-10-31
西部论丛(2020年7期)2020-10-20
食品科学(2018年10期)2018-05-23
好孩子画报(2016年4期)2016-11-19
西南农业学报(2016年5期)2016-05-17
西南农业学报(2016年6期)2016-04-16
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
