
副猪嗜血杆菌荧光定量PCR 检测方法的建立
2012-09-26 00:52:40苗立中沈志强韩文瑜
动物医学进展 2012年11期
苗立中,沈志强,韩文瑜
(1.吉林大学畜牧兽医学院,吉林长春130062;2.山东省滨州畜牧兽医研究院,山东滨州256600)
副猪嗜血杆菌病被称为猪革拉泽氏病,是由副猪嗜血杆菌(Haemophlius parasuis,HPS)感染引起的猪的一种传染病[1]。近年来,随着规模化养猪业的发展,该病已成为影响养猪业的新发典型细菌性疾病[2],给我国养猪业带来了巨大的经济损失。目前,我国对副猪嗜血杆菌病的诊断主要通过传统的病原分离与培养、血清学方法以及常规分子生物学方法。但由于HPS对营养要求苛刻,直接从病料中很难分离到HPS,且该菌培养期间易发生杂菌污染,不易纯化[3]。已有多位学者建立了检测HPS的PCR方法,但是这些常规PCR方法无法对病料中的HPS进行准确定量检测,而且都较繁琐,容易出现假阳性,检测结果具有波动性[4-5]。因此,有必要建立一种新的定量检测技术。
Real-time PCR是在PCR定性技术的基础上发展起来的核酸定量技术,为实时监测PCR扩增并进行定量解析的方法。Real-time PCR具有操作简单、快速、灵敏度高、特异性好及可靠性强的优点,同时又实现了对模板的准确定量,其检测线性范围较宽,不易出现假阳性,在整个反应过程中可自动通过分析荧光信号记录数据,对扩增产物进行精确定量分析,解决了PCR假阳性和溴化乙锭(EB)对环境的污染问题[6],已成为病原学检测的重要手段[7]。
本方法选取HPS的inf B基因中一段高度保守的基因序列作为目的片段,由此设计一对特异性引物,建立HPS基于SYBR Green I荧光染料的荧光定量PCR快速诊断和定量分析检测方法。
1 材料与方法
1.1 材料
1.1.1 菌株 HPS菌株,由山东省滨州畜牧兽医研究院预防兽医学与动物生物技术重点实验室分离、鉴定和保存;大肠埃希菌DH5α、猪胸膜肺炎放线杆菌、猪沙门菌、猪大肠埃希菌、猪巴氏杆菌、猪链球菌等均由本实验室保存。
1.1.2 试剂及试剂盒 p MD18-T 载体、Emerald Amp PCR Master Mix(2×Premix)、SYBR Green Real Time PCR Master Mix(2×)、DNA Marker DL 2 000、EASY Dilution(For Real-time PCR)等均为宝生物工程(大连)有限公司产品;多功能DNA纯化回收试剂盒(离心柱型)、高纯质粒小量制备试剂盒(离心柱型)和细菌基因组DNA提取试剂盒(离心柱型)均为北京百泰克生物技术有限公司产品。
1.1.3 主要仪器设备 紫外可见分光光度计(2000/2000C型)为北京普析通用仪器有限公司产品;Real-time PCR仪(light Cycler480 II型)为德国罗氏诊断产品有限公司产品;直流电泳仪(DYY-8C型)为北京市六一仪器厂生产;BIO-RAD PCR仪为BIO-RAD公司产品;紫外透射反射仪(WD-9403C型)为北京沃德生物医学仪器公司产品。
1.2 方法
1.2.1 引物设计 参照GenBank数据库中 HPS的infB基因的序列(DQ:781933.1),设计一对特异性引物,目的片段长度为129 bp,由上海生工生物工程技术服务有限公司合成。
inf B-F: 5′-AGTAGCAGCTGACGATGGCGTAAT-3′; infB-R: 5′-T CTACACGCTCTGGGTTTGCTTCT-3′。
1.2.2 常规PCR扩增 采用直接水煮沸法从HPS培养液中提取基因组DNA[8],作为常规PCR反应的模板。常规PCR反应体系为25μL,其中PCR Master Mix(2×Premix)12.5μL,去离子水10.5 μL,模板DNA 1μL,引物infB-F、infB-R各0.5μL。反应条件:95℃5 min;95℃30 s,58℃30 s,72℃30 s,30个循环;72℃10 min。反应结束后,取7μL PCR产物于10 g/L琼脂糖凝胶进行电泳鉴定。
1.2.3 质粒标准阳性模板的制备
1.2.3.1 目的基因的纯化回收 将1.2.2中常规PCR产物于10 g/L琼脂糖凝胶电泳后,紫外灯下切回目的片段,用多功能DNA纯化回收试剂盒回收目的DNA,置-20℃保存备用。
1.2.3.2 目的基因的克隆及鉴定 将回收的HPS的infB基因克隆到p MD18-T载体上,然后,将连接产物转化入感受态细胞DH5α中。挑取单个菌落扩大培养,进行菌液PCR鉴定,然后用高纯质粒小量制备试剂盒提取质粒进行PCR及琼脂糖凝胶鉴定,最后将阳性质粒送上海生工生物工程技术服务有限公司测序鉴定。
1.2.3.3 重组质粒浓度的测定 利用紫外可见分光光度计测定阳性重组质粒的浓度,并计算其拷贝数。
1.2.3.4 标准模板的制备 用EASY Dilution(for real time PCR)将阳性重组质粒稀释至1.0×1010copies/μL左右,再依次做10倍梯度稀释,分别以109、108、107、106、105、104、103、102、101、100十个拷贝数梯度作为标准模板。
1.2.4 荧光定量PCR检测方法的建立
1.2.4.1 SYBR GreenⅠ荧光定量PCR方法反应条件的优化 以标准阳性质粒为模板,infB-F/R为引物,选用不同的引物浓度、退火温度、反应时间等进行Real-time PCR反应,根据PCR反应结果,不断优化反应条件,以得到最佳的荧光定量PCR反应条件。
1.2.4.2 标准曲线的建立 以108、107、106、105、104、103、102copies/μL 7个梯度浓度的标准质粒为模板,infB-F/R为引物,进行荧光定量PCR扩增,得出各自的动力学曲线,反应结束后由仪器软件自动分析阈值,结合扩增曲线计算出Ct值,自动拟合生成标准曲线,并计算出曲线的相关系数R2和PCR的扩增效率。
1.2.4.3 熔解曲线的建立 荧光定量PCR反应结束后,由60℃开始做熔解曲线,到95℃结束,连续收集荧光信号,以温度为横轴,以荧光值变化速率为纵轴建立熔解曲线。经软件分析后获得扩增产物的Tm值。
1.2.5 敏感性试验 以107、106、105、104、103、102、101copies/μL的标准品作为模板,进行荧光定量PCR扩增,得出能检测出阳性结果的最低稀释度的拷贝数,同时用常规PCR对以上每个稀释度的标准质粒进行检测,并比较两种方法的敏感性。
1.2.6 重复性试验 选取108、107、106、105、104、103、102copies/μL浓度梯度的阳性标准质粒,每个稀释度一次进行3个重复。根据每个稀释度的Ct值计算变异系数,确定该荧光定量PCR方法的组内重复性。
1.2.7 特异性试验 用细菌基因组提取试剂盒提取大肠埃希菌、巴氏杆菌、胸膜肺炎放线杆菌、沙门菌等猪源性细菌的基因组,再分别取等量的且同等拷贝数的样品DNA,以其为模板,inf B-F/R为引物,以25μL体系且相同条件进行荧光定量PCR检测。
2 结果
2.1 目的基因的克隆与鉴定
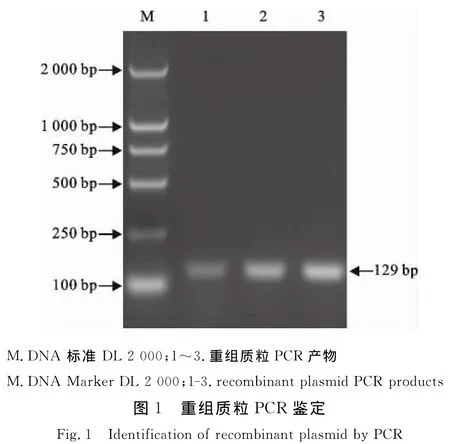
PCR产物经回收后,与p MD18-T载体连接构成重组质粒,用1.2中的引物对所构建的质粒进行常规PCR扩增,得到一条129 bp左右的DNA片段,与连接前的PCR产物大小一致(图1)。将经PCR鉴定的阳性重组菌送上海生工生物工程技术服务有限公司进行序列测定,测定结果与目的片段序列完全一致。
2.2 HPS荧光定量PCR检测方法的建立
2.2.1 荧光定量PCR反应条件的优化 优化后的荧光定量PCR反应体系为25μL,其中SYBR Green real-time PCR Master mix(2×)为12.5μL,去离子水10.5μL,inf B-F、inf B-R 各0.5μL,标准模板1μL。荧光定量PCR反应条件:95℃3 min;95℃15 s,58℃30 s,72℃30 s,40个循环;在72℃收集荧光;95℃15 s,60℃30 s,95℃ 建立熔解曲线,1个循环;40℃10 s。
2.2.2 标准曲线的建立 按照以上最佳的优化条件,选 择 108、107、106、105、104、103、102copies/μL 7个梯度浓度的标准质粒作为模板,制作动力学曲线(图2)和标准曲线(图3)。动力学曲线的扩增效率为1.951,标准曲线斜率为-3.444,纵轴截距为36.63,线性方程为 Y=-3.444X+36.63,相关系数为0.998 8,表明线性关系很好。

图2 荧光定量PCR扩增动力学曲线/(copies·μL-1)Fig.2 Dynamic curve of fluorescent quantitative PCR(copies·μL-1)

图3 荧光定量PCR标准曲线Fig.3 Standard curve of fluorescent quantitative PCR
2.2.3 熔解曲线的分析 熔解曲线分析可以区分非特异性扩增,以验证PCR产物的特异性。由图4可以看出,本试验的熔解曲线是单峰,表明引物的特异性很好,没有出现非特异性扩增和引物二聚体。

图4 SYBR GreenⅠ荧光定量PCR熔解曲线Fig.4 Melting curve of the SYBR Green I real-time PCR
2.3 敏感性试验结果
通常我们认为,模板的起始数越低Ct值则越大,当Ct值达到35以上时,定量检测结果是不准确的。试验结果显示,实时荧光定量PCR能检测出的模板最低浓度为10 copies/μL(图5),而常规PCR能检测出的最低模板浓度为1.0×103copies/μL(图6),这表明荧光定量PCR的敏感性比常规PCR高100倍。

图5 荧光定量PCR敏感性试验/(copies·μL-1)Fig.5 Sensibility test of fluorescent quantitative PCR(copies·μL-1)

图6 常规PCR敏感性试验/(copies·μL-1)Fig.6 Sensibility tests of routine PCR/(copies·μL-1)
2.4 重复性试验结果
试验选 取 108、107、106、105、104、103、102copies/μL的标准模板以优化的反应条件分别连续扩增3次,结果显示,每个梯度的样本重复性很好(图7)。同时,对检测结果进行统计学分析,结果表明,组内重复性试验变异系数小于2.5%,说明该方法具有较好的重复性与稳定性(表1)。

表1 荧光定量PCR的组内重复性试验结果Table1 Intra repeatability test of fluorescent quantitative PCR

图7 荧光定量PCR重复性试验Fig.7 Repeatability test of fluorescent quantitative PCR
2.5 特异性试验结果
以猪胸膜肺炎放线杆菌、猪沙门菌、猪大肠埃希菌、猪巴氏杆菌的DNA,进行SYBR GreenⅠ荧光定量PCR反应,结果这些细菌的扩增曲线始终没有出现上扬,几乎为水平线,表明该方法有很好的特异性。
3 讨论
在荧光定量反应中,16 SrRNA基因不能鉴别HPS和某些同源性细菌,而inf B基因在种属间的变异率较低且兼顾有不同血清型之间的保守序列[9],因此本试验选取HPS的inf B基因中的一段高度保守的序列作为目的片段,由此保证了该检测方法的通用性。
SYBR GreenⅠ染料可以与双链DNA分子的小沟结合,其荧光强度的增加与双链DNA的数量成正比,从而保证了荧光信号的增加与PCR产物的增加完全同步,以达到进行实时定量的目的。由于SYBR GreenⅠ染料与双链DNA的结合没有特异性,容易产生非特异性扩增[10],因此,试验需要对荧光定量PCR反应条件进行优化以增强检测的特异性和准确性。本试验通过反应条件的优化,在退火温度为58℃,获得的熔解曲线只有1个特异性峰值,表明该反应为特异性扩增,没有出现引物二聚体和其他非特异性的扩增。
试验结果表明,实时荧光定量PCR方法运用于检测副猪嗜血杆菌具有快速、高效、灵敏的优点,并且实现了对模板的准确定量。该方法检测的线性范围较宽,因此不易出现假阳性,在整个反应过程中可随时对扩增产物进行精确定量分析,具有无污染性和实时性等优点,解决了常规PCR检测方法中存在的费时费力、容易污染及检测结果波动性的问题。因此,实时荧光定量检测副猪嗜血杆菌技术具有广阔的应用前景。
[1]刘俊伟,安志兴,葛亚明,等.副猪嗜血杆菌病的病原分离鉴定与病理学诊断[J].黑龙江畜牧兽医,2010,12(1):111-113.
[2]司振书,王桂英.副猪嗜血杆菌病研究进展[J].中国畜牧兽医,2011,38(6):179-182.
[3]张培君,孙惠玲,苗得园,等.猪胸膜肺炎放线杆菌和副猪嗜血杆菌的鉴别诊断及血清型鉴定[J].中国兽药杂志,2002,36(10):18-20.
[4]Ruiz A,Oliveira S,Torremorell M,et al.Outer membrane proteins and DNA profiles in strains of Haemophilus parasuis recovered from systemic and respiratory sites[J].Clin Microbiol,2001,39(5):1757-1762.
[5]Jung K.Development of polymerase chain reaction and comparison with in situ hybridization for the detection of Haemophilus parasuis in formalin-fixed,paraffin-embedded tissues[J].Vet Med Sci,2004,66:841-845.
[6]宋修庆,高宏雷,王晓艳,等.鸡贫血病毒Taq Man探针荧光定量PCR检测方法的建立[J].中国预防兽医学报,2009,31(1):56-59.
[7]母安雄,应小飞,陶 益,等.副猪嗜血杆菌的研究进展[J].中国畜牧兽医,2005,32(11):48-49.
[8]蔡旭旺,陈焕春,刘正虼,等.副猪嗜血杆菌的分离培养和血清型鉴定[J].华中农业大学学报,2005,24(1):55-58.
[9]于 江,吴家强,张玉玉,等.副猪嗜血杆菌荧光定量PCR检测技术的建立与应用[J].家畜生态学报,2010,31(4):77-81.
[10]叶晓艳.实时荧光定量PCR检测肠道病毒方法的建立[D].湖北武汉:华中科技大学,2009.
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中成药(2018年12期)2018-12-29 12:25:44
食品科学(2018年10期)2018-05-23 01:27:28
中成药(2017年6期)2017-06-13 07:30:35
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
西南医科大学学报(2015年1期)2015-08-22 13:01:46
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
医学研究杂志(2015年4期)2015-06-10 06:42:43
