
价层电子对互斥理论教学中几个问题的讨论
2012-09-25 03:35张晨曦王雪峰
大学化学 2012年4期
张晨曦 王雪峰
(同济大学化学系 上海 200092)
在大学化学中的无机化学及结构化学课程中,价键理论体系是学习的基础也是教学的重点。在众多价键理论中,价层电子对互斥(VSEPR)理论基于简单的分子Lewis结构式进行推测,对于大部分分子构型的预测与解释准确而简便。因此,VSEPR理论已经同经典价键理论、杂化轨道理论以及分子轨道理论一并成为大学化学价键理论体系的核心内容。
然而,通过多年的教学和学生的反馈,并对现今许多无机化学和结构化学教材进行对比后,发现在VSEPR理论教学中还存在以下3方面问题:
① 对于价层电子对产生相互排斥作用的原因未能给出明确的说明;
② 虽然明确指出VSEPR理论不适用于过渡金属化合物构型的预测,但未能给出合理的解释;
③ 学生未能清楚地理解VSEPR理论与杂化轨道理论的本质区别,误认为VSEPR理论所预测的分子构型就是相应中心原子的杂化轨道构型。
在此,笔者试图通过对以上问题的讨论,让学生更深刻地理解VSEPR理论的理论基础,同时将现今研究前沿的不规则过渡金属化合物放入价键理论体系中讨论,希望能对今后VSEPR理论的教学有所裨益。
1 VSEPR理论的理论基础
在如今的许多教材以及教学过程中,对价层电子对相互排斥的原因都含糊其辞,仅仅指出价层电子对应该在空间尽量地远离,但都未能给出合理的解释[1-5]。这就会使一些学生想当然地认为,价层电子对相互排斥是因为它们之间的静电排斥作用。这成为许多学生在学习VSEPR理论时的一个误区。其实,早在Ronald J.Gillespie提出VSEPR理论时就已明确指出,价层电子对相互排斥的主要原因在于Pauli效应产生的相互排斥力而不是它们之间的静电斥力[6]。
根据Pauli不相容原理,自旋相反的电子倾向于在一起,电子的这种相关效应叫Pauli效应;而库仑斥力可表述为静电效应,这种作用力使电子彼此分离。Pauli不相容原理是在经典价键理论中提出的,其实Pauli效应是电子在原子或分子体系中的一种基本性质[7]。由Pauli效应而产生的斥力主要是自旋同向电子间轨道回避的量子效应,是一种近程相互作用[8]。也就是说,Pauli不相容原理不仅仅适用于自旋相反的单电子形成价键,对于所形成的价键也可看作是“同性”的。根据Pauli不相容原理,这种“同性”的价键也应该相互排斥,在空间上表现为呈几何上相距最远的构型。这种Pauli斥力与自旋同向电子间距离的8~10次方成反比,相比之下,静电斥力与同性电荷之间距离的2次方成反比[8]。这就表明,在中心原子周围很小的范围内,Pauli斥力要比静电斥力更重要,是价层电子对相互排斥的主要原因。由于随着距离的增大,Pauli斥力急剧下降,因此,只有在价层电子对这样的近程作用时才提及Pauli斥力,当相互作用距离大一些时,静电斥力就成了考虑的主要因素。
由于平时很难接触到如此小范围内的相互作用,因此很多学生甚至教师都会误认为价层电子对之间的相互排斥来自静电斥力。由上述论述可以知道,价层电子对互斥理论有其严谨的理论基础。虽然Pauli斥力的大小很难精确计算和测量,并且Pauli斥力与静电斥力是协同关系。但是,我们不能把静电斥力与Pauli斥力混为一谈。我们建议,为了学生能够更深刻地理解价层电子对相互排斥的原因,在今后的无机化学教材中能够补充上述内容,这也可以使教材对VSEPR理论的介绍更为完善。教师在教授这部分内容时,可以详细地介绍一下Pauli斥力,让学生明白Pauli不相容原理是在近程作用中广泛适用的基本性质。
2 过渡金属不适用于VSEPR理论的原因
通常,教材在介绍VSEPR理论的最后会指出,该理论对于过渡金属化合物分子构型的预测往往和量子计算以及实验现象不符,但是没有能够给出不符合预测构型的例子以及VSEPR理论“失效”的合理解释[1-5]。随着对过渡金属化合物的研究不断深入,尤其是金属有机领域不断发展,越来越多不符合VSEPR理论预测的化合物相继被发现。如今,这样的不规则化合物已在材料科学、催化以及金属有机领域成为研究的前沿。因此,对VSEPR理论不适用于过渡金属化合物构型原因的探究,不仅可以让学生进一步了解VSEPR理论的局限性,同时也可以将科学研究的前沿纳入到经典价键理论的教学中。
在不符合VSEPR理论预测的分子构型中,六配体过渡金属氢化合物的不规则构型是当今研究的热点。下面以我们在实验室所观测到的现今最高金属氢化物WH6为例(图1),解释VSEPR理论不适用于过渡金属化合物的原因。
根据VSEPR理论,价层电子对数目为6的WH6,其构型应该是正八面体(Oh)。但是,经量子化学计算以及实验观察[9],WH6呈上方开口较大,下方开口较小的畸变三角锥构型(C3v)。显然,仅仅通过VSEPR理论中价层电子对的相互排斥作用不能合理解释产生这样不规则构型的原因。
既然VSEPR理论能够成功地对主族元素化合物的构型进行预测和解释,那么我们就可以从不同的方面入手进行对比分析。相比主族元素,过渡金属有d轨道参与轨道杂化成键。就WH6来说,中心原子W相对于S来说,W的杂化方式为sd5而S最多为sp3d2杂化。在经典价键理论中有最大重叠原理,即成键电子的原子轨道在对称性一致的前提下发生重叠,原子轨道的重叠程度越大,两核间电子的概率密度就越大,形成的共价键就越稳定[1]。依据该理论,配体在与中心原子成键的过程中,为使得成键最稳定,必定采取和中心原子杂化轨道重叠最大的方向成键。由于s轨道为球形,配体在任意方向都可与其最大程度重叠成键。因此,sdn杂化轨道更多的是体现d轨道的形状。由于d轨道呈“花瓣状”,相比于直线型的p轨道来说,其杂化轨道不是常见的规则形状。根据相应的量子计算,为满足d轨道最大程度地参与成键,sd5杂化轨道的形状为畸变三角锥型(C3v)[10]。同时,相应的分子轨道模型也得出了同样的结果[11]。由于WH6分子中H配体在与中心原子成键后,不存在孤对电子的排斥作用。所以其分子构型能够很好地显示其中心原子sd5杂化轨道的原貌。
从上述分析可知,VSEPR理论之所以不适用于过渡金属化合物构型的预测,是因为d轨道参与成键后,其中心原子杂化轨道与常见的p轨道参与形成的杂化轨道不同。对过渡金属不规则构型的讨论分别涉及了VSEPR理论、杂化轨道理论、分子轨道理论以及经典价键理论中的最大重叠原理,这样可使学生既能了解现今研究的前沿领域,又能将整个基础价键理论体系有机地联系在一起。我们建议在无机化学及结构化学教材中,将上述事例作为课外拓展,这样既可以将经典理论的学习和科学研究的前沿结合起来,也可以提高学生利用所学基础理论知识解决实际问题的能力。在教师讲授VSEPR理论的过程中,也可以使用这个例子作为对整个价键理论综合运用的思考题。这符合教学与科学研究前沿相结合这一教学改革的方向。
3 VSEPR理论与杂化轨道理论的关系
在价键理论的教学过程中,由于杂化轨道理论和VSEPR理论几乎是同时讲授的,因此很多学生容易将VSEPR理论和杂化轨道理论混淆,不能分辨VSEPR理论与杂化轨道理论的本质区别。另外,教师在教授学生运用价键理论解释分子构型时,常常让学生先用VSEPR理论预测分子构型然后通过杂化轨道理论解释,一些教材中的习题也是这样设计的[1]。这样虽然对于大部分主族元素能够得出正确的结论,但也更容易使学生将两种理论混为一谈,同时产生VSEPR理论预测的分子构型就是相应中心原子杂化轨道形状的误解。
例如用VSEPR理论预测CCl4的分子构型,价层电子对数目为4,所以该分子构型为正四面体,再利用杂化轨道理论得出sp3杂化轨道也为正四面体,这也符合量子化学计算结果以及实验的验证。
然而,使用VSEPR理论预测WF6的分子构型,可得其价层电子对数目为6,呈正八面体构型(图2),这和量子化学计算以及实验结果完全一致[12]。但如上述分析可知,中心原子W的sd5杂化轨道形状是C3v的畸变三角锥而不是正八面体构型。由此可见,VSEPR理论所预测的分子构型和相应中心原子杂化轨道形状是不相同的。
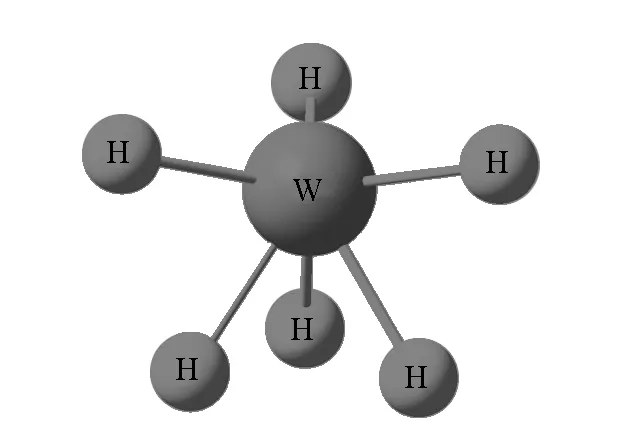
图1 WH6的畸变三角锥构型(C3v)

图2 WF6的正八面体构型
究其原因,是由于VSEPR理论和杂化轨道理论有本质上的区别。VSEPR理论是忽略许多次要因素,抓住价层电子对相互排斥这一主要因素,结合简单的几何知识对整个分子的构型进行预测。该理论不考虑中心原子的杂化轨道的形状,仅仅考虑价层电子对的数目,多用于对分子构型的预测。而杂化轨道理论是对经典价键理论的拓展,不涉及整个分子的构型,而是着眼于中心原子与配体成键的过程。通常运用杂化轨道理论是对已知的分子构型从成键的角度进行解释,而不是用来预测分子的构型。对于WF6分子来说,其分子构型除了受中心原子杂化轨道形状的影响外,还受F配体的排斥作用,这就使得WF6正八面体构型(图2)远远偏离其中心原子杂化轨道的C3v形状。
从上述分析可以看出,VSEPR理论和杂化轨道理论既有紧密的联系又有本质的区别。在教学过程中,学生很容易混淆它们的概念。因此,我们建议在相关习题设计时,可以在解答过程后对其进行说明,防止学生产生误区。另外,教师在教授这部分内容时,应该在分析各个价键理论的基础上,再指导学生综合地运用整个价键理论体系解决问题。
4 结语
大学化学无机化学及结构化学课程中的价键理论是本科生日后进一步学习化学的基础,而VSEPR理论以其简单有效的特点越来越受到学生的青睐。本文着重论述了VSEPR理论的理论基础、不适用于过渡金属的原因以及该理论和杂化轨道理论的区别,并提出一些针对VSEPR理论教学以及教材编写的建议,希望能使学生对VSEPR理论有更深入的理解。
[1] 宋天佑,程鹏.无机化学(上).北京:高等教育出版社,2004
[2] 黄可龙.无机化学.北京:科学出版社,2007
[3] 天津大学无机化学教研室.无机化学.第4版.北京:高等教育出版社,2010
[4] 宋天佑.简明无机化学.北京:高等教育出版社,2007
[5] 古国榜,李朴.无机化学.第2版.北京:化学工业出版社,2007
[6] Gillespie R J.JChemEduc,1970,47:18
[7] 严成华.化学通报,1977(4):60
[8] 郭子仪.大学化学,1988,3(6):16
[9] Wang X,Andrews L.JAmChemSoc,2002,124:5636
[10] Kang S K,Tang H,Albright T A.JAmChemSoc,1993,115:1971
[11] Landis C R,Firman T K,Root D M,etal.JAmChemSoc,1998,120:1842
[12] Nakamoto K.Infrared and Raman Spectra of Inorganic and Coordination Compounds.6th ed.New Jersy,USA:A JOHN WILEY & SONS INC,2009
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
原子与分子物理学报(2020年5期)2020-03-17
陕西中医(2018年6期)2018-08-29
考试周刊(2018年39期)2018-04-19
北京航空航天大学学报(2017年10期)2017-04-20
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
中国塑料(2016年11期)2016-04-16
