
Ⅰ型聚酮类天然产物中利用点突变产生类似物的研究
2012-09-21 09:16刘伟高菊芳
浙江化工 2012年7期
刘伟 高菊芳
(上海师范大学生命与环境科学学院,上海 2000234)
生物化工
Ⅰ型聚酮类天然产物中利用点突变产生类似物的研究
刘伟 高菊芳
(上海师范大学生命与环境科学学院,上海 2000234)
点突变是指通过聚合酶链式反应(PCR)等方法向目的DNA片段中引入所需变化(通常是表征有利方向的变化),包括碱基的添加、删除、改变等。通过构建发生突变的质粒利用遗传转移的方法同源重组到野生型基因组中,使得氨基酸改变从而引起蛋白功能发生变化,获得鉴定正确的突变株后发酵筛选结构类似物。聚酮类天然产物在生物合成过程中,其聚酮链会发生若干步氧化还原反应,通过点突变的方法失活该蛋白,从而跳过该步氧化还原反应的作用,产生PKS骨架上的改变,从而得到结构类似物。
聚酮类化合物;点突变;结构类似物
聚酮类天然产物是一大类以乙酰或丙二酰单酰辅酶A为延伸或者起始模块,在聚酮合成酶(polyketide synthase,PKS)催化下经过多轮克莱森缩合(Claisen缩合)和高度修饰后形成的天然产物,许多细菌尤其是放线菌、真菌甚至植物都具有形成此类天然产物的能力,一般来讲,该合成阶段都发生在次级代谢阶段,有些甚至不是其生长繁殖所必需[1-2]。
这一类天然产物通常具有诸如红霉素等抑制细菌、两性霉素等抑制真菌以及阿霉素等控制癌症的活性,广泛运用于医药、农业、畜牧业等。其良好高效的生物学活性、巨大的新药开发潜质、还有独特的酶学机制以及蛋白间的相互识别作用无不吸引和推动大家的重视与研究。由于其精密而又经济的合成机制和调控机制,越来越多的分子生物学手段在其结构改造上得以实现,这无论是对于对抗由于细菌耐药性广泛的出现而引起的抗生素受限还是揭示新的非化学合成的各种反应都具有巨大的意义。本文关于聚酮合酶的操作就是利用组合生物合成的方法得到了该天然产物的类似物[3]。
聚酮化合物是通过聚酮合酶(polyketide sythase,PKS)催化形成,目前已知的聚酮合酶分为三大类,分别为Ⅰ型,Ⅱ型和Ⅲ型PKS。
Ⅰ型PKS是一个由许多功能域(domain)组成的蛋白,其中至少需要以下三个功能域,即酰基转移酶(acyl tranferase,AT)、酰基载体蛋白 (acyl carrier protein,ACP)和酮基合成酶(ketosynthase,KS),由KS催化AT上的羧酸起始单元或者延伸单元缩合。其次有些非关键的功能域 (并非所有的蛋白中均含有),包括脱水酶 (dehydratase,DH)、烯醇还原酶(enyol reductase,ER)、酮基还原酶(ketoreductase,KR),中途可以负责聚酮链的修饰、脱水或者还原。最后在硫酯酶(thioesterase,TE)的解离下从PKS上脱落,然后利用后修饰基因对其进行进一步的催化,比如环化、糖基化等过程,形成最后的产物。
Ⅱ型PKS是一个多功能酶复合体,只包含一套可以重复使用(interative)的结构域,每一结构域在重复的反应步骤中多次用来催化相同反应,包含了KSα,KSβ,ACP,其中除ACP为酰基载体蛋白以外,通常认为KSβ与KSα不仅负责聚酮链延伸和小分子羧酸的缩合,而且负责聚酮链的链长控制。
Ⅲ型PKS比较特殊,它是一类直接利用酰基-COA上的羧酸进行缩合,一般不需要酰基载体蛋白上的磷酸泛酰巯基乙胺[3-4]。
随着细菌抗药性的产生,市场对于高效低毒的新型化合物的需求,以及化学合成上的困难,对于筛选结构类似活性更高的天然产物类似物,就有了更大的挑战。传统意义上的物理诱变筛选因其工作量大,效果不明显等缺点已经不能够满足对于良好活性类似物的要求。因此科学家们开始在对其生物合成途径中的酶进行选择性的失活来产生新化合物。本文旨在介绍利用点突变的方式使得蛋白功能缺失,从而使得天然产物在其合成过程中跳过该蛋白的催化步骤,在立体结构或者骨架基团上产生变化,产生"非天然"的天然产物,以期获得更好更高效的化合物[5]。
点突变是研究蛋白质结构与功能关系以及确认结构域或特定氨基酸所处位置的重要手段,一般用来调节基因的表达和对DNA分子的修饰。同时还可以通过点突变改变氨基酸残基来引起目标蛋白的失活,导致其功能缺失。在Ⅰ型PKS中利用点突变来跳过或者失活某目标蛋白的催化功能,从而产生与该天然产物结构类似(如羟基保留)的产物[6]。其主要操作方法包括重叠延伸法、扩增质粒全长法、试剂盒等[7-8]。
聚酮合酶中还原型功能域的点突变失活较为常用的方法为重叠延伸法,其主要原理为利用PCR的方法引入所需突变(将突变碱基设置在引物中),成功后利用大肠杆菌与链霉菌等的同源重组构建至野生型菌株(结合转移,电转化等方式),同时利用同义突变引入酶切位点进行验证,加上测序等手段检测到成功突变的突变株。进一步发酵检测寻找目标产物[9]。
1 格尔德霉素类似物的产生
格尔德霉素(Geldnamycin)是一类苯醌类大环内酯,因其可以竞争性的抑制热激蛋白,所以曾被考虑作为抗癌药物,但是由于其肝毒性阻止了进一步的临床应用,于是就启发人们去寻找毒性降低的类似物。其中17-丙酰基氨基-17-去甲氧基格尔德霉素已经在进行一期、二期临床试验。
格尔德霉素由吸水链霉菌NRRL3602产生,它是通过一个3-氨基-5-羟基-苯甲酸(AHBA)的前体装载至前体合成功能域上,经过三个PKS酶的催化,完成了七步的延伸,形成了聚酮链,随后在解离后通过一系列的后修饰,最终形成完整的分子[10-11]。
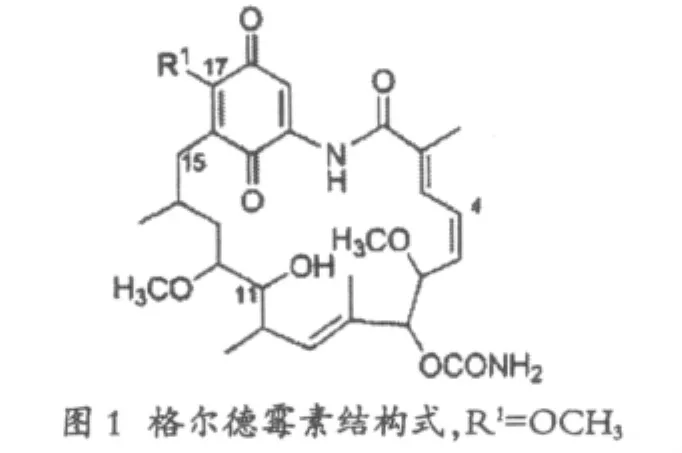
韩国科学家Young-Soo Hong等[12]利用对PKS酶中个别功能域的点突变,选择格尔德霉素生物合成基因簇中gelA基因中的DH1功能域,将其催化中心的HIS残基定点突变,产生功能缺失的脱水酶,因此保留了格尔德霉素15位的羟基,利用PCR的方法引入关键氨基酸的失活,然后利用大肠杆菌和链霉菌的同源重组,发生两次交换后废除了蛋白的功能。
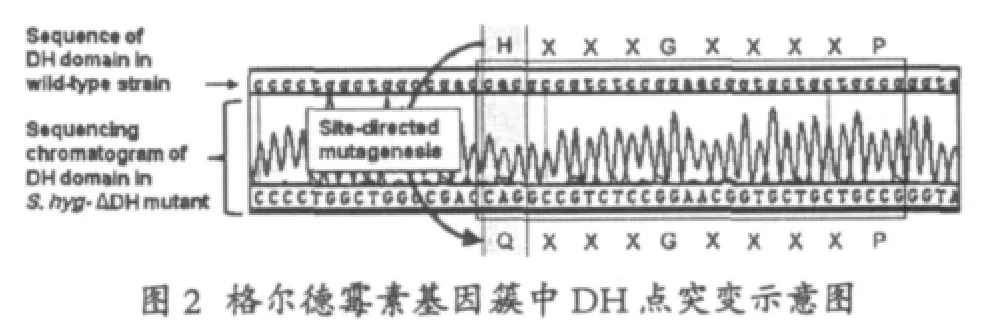
通过抗性筛选到突变成功的突变株,发现产生了新的化合物,由于DH的失活,使得在聚酮链上的羟基得以保留,最终检测发现,根据MS分子量推断,该化合物正是15位保留羟基的格尔德霉素衍生物,这一化合物成功检测到后,该课题组利用同样的操作方法对聚酮合酶进一步进行选择性的失活,通过失活基因簇中相应的后修饰基因获得了下列的类似物:
DHQ1:除了因DH5失活而得以保留的羟基外,将gel16进行失活,从而使得C4,C5之间的双键饱和,并且负责17位后修饰基因gel16失活,gel16负责将格尔德霉素中的C4,C5间的双键的形成,产生DHQ1,并且由于C15位与C11位发生了脱水形成一个环氧的结构,DHQ2为保留有C4,C5间双键的DHQ1,对于基因簇中的后修饰基因gel7突变,产生了C-15位羟基,C-17位去甲氧基的格尔德霉素类似物DHQ3,对于基因簇中gel7和gel8进行双突变,得到了DHQ4,即去甲酰基的DHQ3。
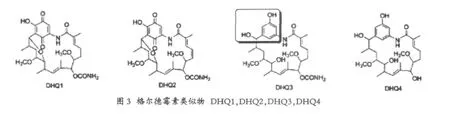
利用酵母Hsp90 ATP酶含量测定的方法来对图3的化合物进行活性测定(见表1)。
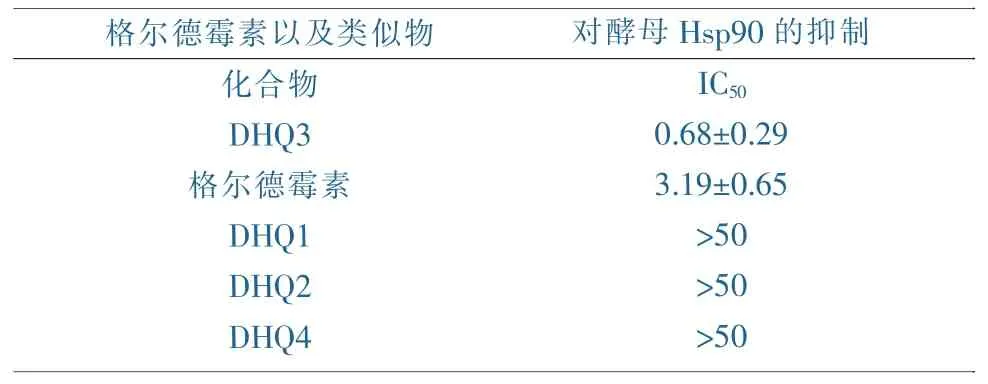
表1 格尔德霉素以及类似物的活性测定
其中发现15位羟基,17位去甲氧基格尔德霉素DHQ3相对于原来的格尔德霉素,活性提高了将近5倍,而其余的三个类似物没有活性[12]。
2 制霉菌素类似物的产生
制霉菌素A1(NystatinA1)是一类多烯类大环内酯广谱抗真菌药,由诺尔斯链霉ATCC11455产生,两性霉素B(Amphotericin B)是其较著名的类似物,与制霉菌素A1差别不大,一是在C28~C29位的差别,前者是一个双键,而制霉菌素A1则为饱和键,二是两性霉素中C8位上含有羟基,C10位为饱和键,而制霉菌素相反[13]。
2005年挪威科学家揭示了制霉菌素的生物合成过程,它由一个乙酸单位起始,经过18次的延伸,每次装载一个二碳单元,最后在硫酯酶的作用下水解离开聚酮合成酶,形成一个三十八元大环内酯,聚酮合成酶中的还原性功能域催化形成了大环内酯上的烯基,之后再经过后修饰基因对于骨架进行进一步的催化,最后形成制霉菌素A1[14]。不同构造的还原型功能域和其不同活性功能对于最后骨架上的基团一一对应,例如在KR,KR+DH,KR+DH+ER分别存在的情况下会产生制霉菌素骨架上的羟基,双键以及饱和单键[15]。
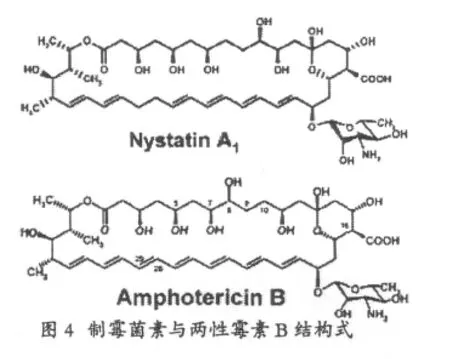
由于制霉菌素的溶血毒性及可溶性等问题,限制了其在人系统性真菌病中广泛应用,所以对于其活性类似或者更高但毒性减少甚至消失的类似物的发现和制备提出了要求。
S44HP是2004年由Bruheim等人通过对制霉菌素生物合成基因簇Nysc蛋白中的烯醇还原酶5(ER5)点突变失活,成功筛选得到发酵突变株,发酵检测得到了C28~C29位烯基保留的制霉菌素的类似物,其抗菌活性与两性霉素相当,比制霉菌素的活性要高。可能的原因是因为在结构上更加类似。但是可溶性是两性霉素的10倍左右。
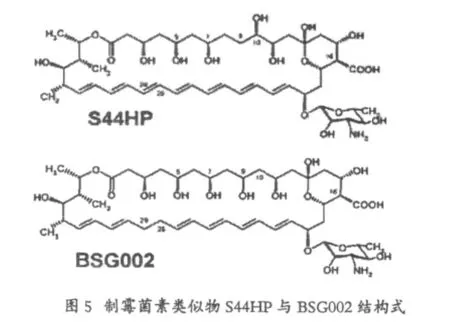
2008年,Sergey B Zotchev等人利用点突变的方法将NysJ中ORF15的脱水酶功能域(DH15)中催化残基His失活,使得C9位上的羟基得以保留,得到了类似物BSG002,由于这一改变,使得后修饰基因NysL不能正确的识别原先C9位并没有羟基的结构,因而不能执行其对于C10位的羟化反应,活性测试表明,与制霉菌素A1相比,BSG002虽然在其溶血活性上至少降了两倍,但是同时,其抗真菌的活性也降到了制霉菌素A1的四分之一。由于该突变株的基因型发生了改变,引起了BSG002产量的下降,在同等发酵条件下,仅有野生株产制霉菌素产量的30%左右。
NysN负责的是C16位的氧化,该基因编码一个假定的P450单加氧酶,序列分析发现其保守氨基酸Cys346可能是血红素的结合位点,因此利用点突变的方法将其突变为Ala,产生突变株CL346AS,发酵检测发现该突变株产生了16-去羧基-16-甲基制霉菌素(16-DecNys),体外活性测试显示,其溶血活性下降了约两倍的同时,抗真菌活性几乎没有发生变化。也就是说,由于该氧化酶的失活,使得16位羧基被甲基替换,因而降低了制霉菌素的毒性。同时突变株CL346AS合成16-DecNys的产量非常低,仅有同等发酵条件下制霉菌素产量的2%。据推测可能存在有未知的反馈抑制。
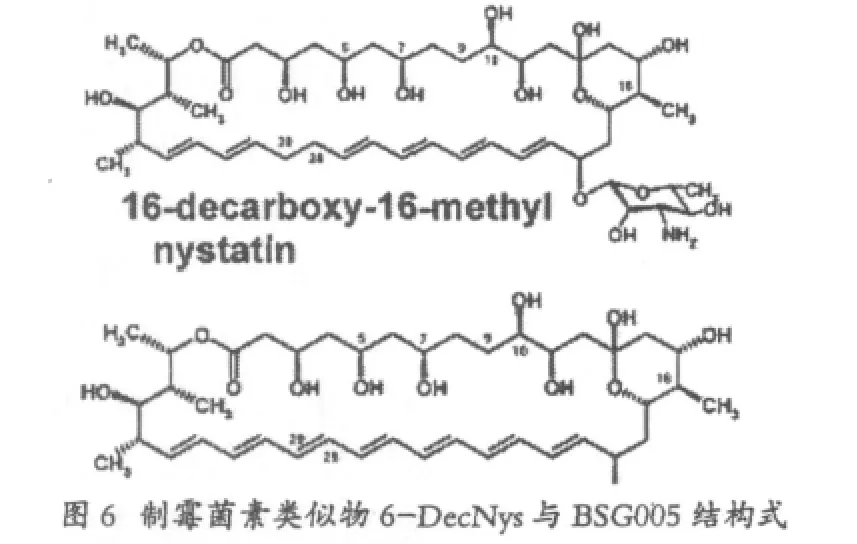
在S44HP的突变株的基础上,同样将 NysN基因失活,得到了16-甲基-28-29脱氢的制霉菌素类似物BSG005,抗菌活性比S44HP提高了近两倍,比制霉菌素A1提高了20倍。而且与16-DecNys相比,产量提高了20倍。
同样,在S44HP的突变株中,失活NysJ中酮基还原酶16(KR16)和酮基还原酶17(KR17)中活性位点Tyr,使得在制霉菌素A1多元醇的区域中C7和C5位的羟基被酮基所取代,得到相应的类似物BSG017和BSG013,活性测试发现与S44HP相比,BSG013的抗真菌活性大约降低了两倍,而溶血毒性有大约1.2倍的提高,而BSG017的抗真菌活降到S44HP的四分之一,溶血毒性大约1.3倍的提高。其产生菌株的产量与S44HP产生菌相比,均下降为其56%与49%。
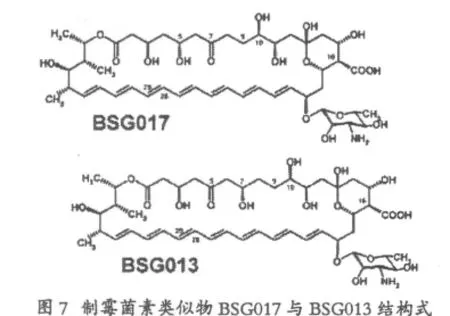
接着作者利用BSG017和BSG013的突变株,将NysN基因失活,进一步得到16位脱羧基甲基取代的 BSG017和 BSG013,命名为 BSG031和BSG020,即为5-酮基-16-甲基-28-29脱氢-制霉菌素A1,7-酮基-16-甲基-28-29脱氢-制霉菌素A1。活性测试表明 BSG031和 BSG020分别比S44HP的溶血毒性更大程度的降低,而抗菌活性得到了提高。产量相应也下降为S44HP产生菌的20%与29%。
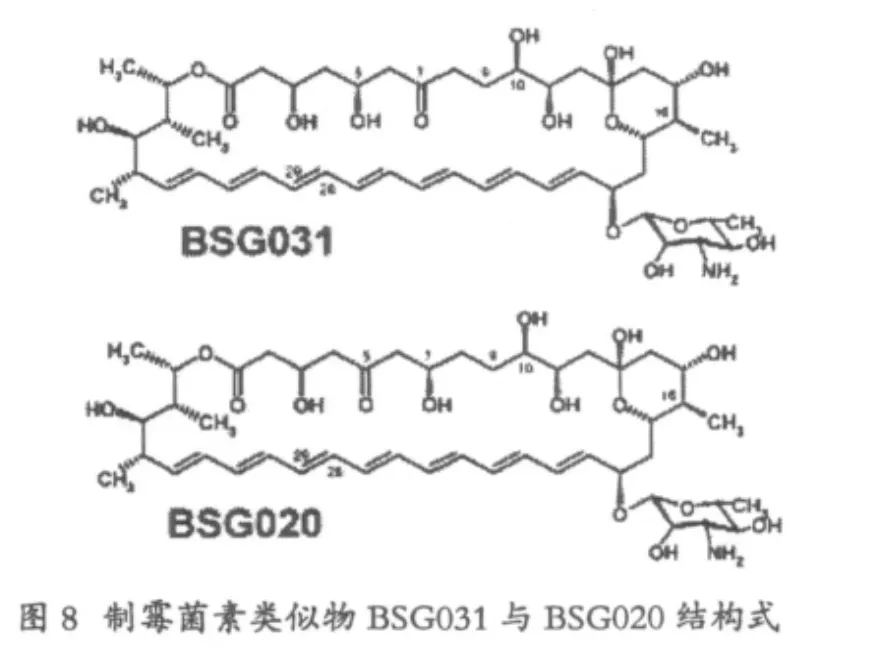
上述类似物活性汇总,以白色念珠菌ATCC10231为测活菌株,以马血红细胞为出发细胞株作为溶血活性测试品株。体外测活结果如表2。
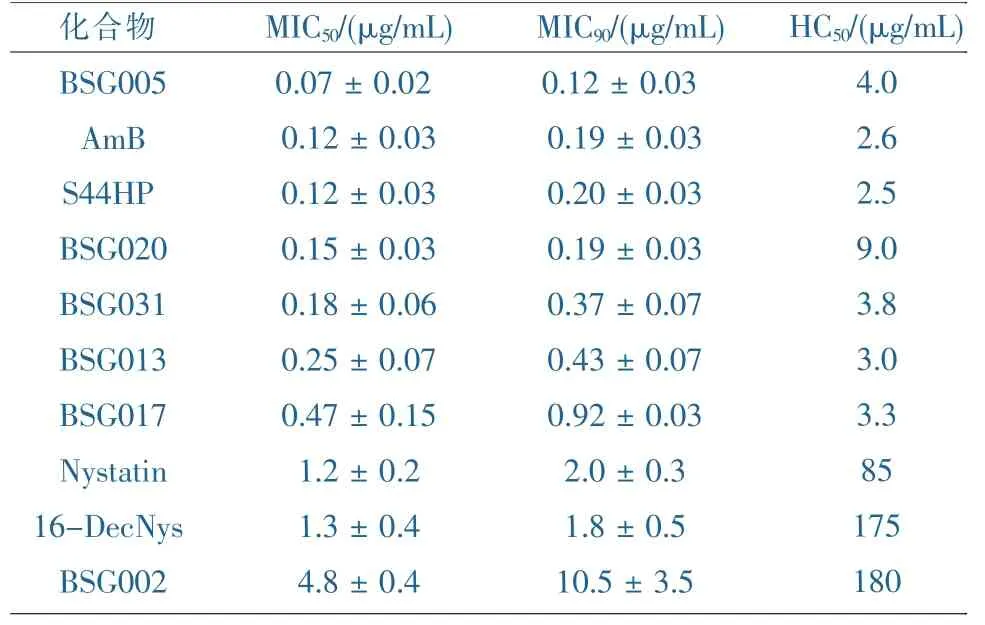
表2 通过基因工程获得的制霉菌素类似物抑菌活性与溶血毒性测试
同时对于制霉菌素和两性霉素B以及类似物S44HP、BSG002、BSG005、BSG020进行被感染大鼠中体内测活,其结果与体外测活均一致[16]。
3 小结
由于Ⅰ型PKS中各个还原型结构域的存在程度不同,通过点突变这一方法,失活这些对于最终的天然产物骨架进行氧化还原的功能域,因而产生大量的结构类似物,这些改变的基团对于最终产物的溶解性、毒性、抗菌活性等至关重要。与传统的物理诱变筛选,点突变这一方法有着明显的优势:
(1)快捷直接。由于分子生物学的飞速发展,通过在基因水平直接对于基因的修饰和改变,产生蛋白功能的变化,合理的设计加上日趋成熟的操作,与原先比较盲目、大海捞针似的筛选相比,更加的经济快速。可以同时尝试更多的结构变化体外测活,从而得到更高效更安全的化合物。
(2)由于微生物体中具有很多经济但复杂的催化机制,通过对其酶蛋白序列的分析和改变,对于很多化学家感兴趣的体内经典或独特的催化反应进行了补充。
但同时,这一方法也有很多的不足之处,最常见的便是产生的突变株产量严重的下降,例如本文中提到的格尔德霉素类似物DHQ1、DHQ2,因为基因型的改变,这些突变株在和野生型同样的发酵条件下,产量有近10倍的减产,再如制霉菌素的研究中,虽然突变株发酵检测到BSG020和BSG031提高了抗菌活性而且其毒性有了大大的降低,但是其产量仅为野生型的20%和29%。究竟为什么基因型的某个氨基酸改变,产量就会有如此显著的差别,还没有研究透彻,但是也有可能是产生的类似物对于菌株生长的不利或者是参与到了复杂而又精密的反馈调节中。另一方面,由于微生物来源广泛,对其基因型进行操作存在着比较明显的差异,常用的遗传转移系统在一些特殊来源或者对于生长状态要求较为特殊的菌株并不奏效,但随着生物技术的愈发成熟,相信点突变这一年轻而又古老的技术在遗传改造和化合物筛选中愈发举足轻重。
[1]Leonard Katz.Manipulation of modular polyketide synthases[J].Chem.Rev.,1997,97:2557-2575.
[2]许杨,魏康霞.真菌聚酮合酶基因的研究进展 [J].食品与生物技术学报,2008,3,27:2,1-5.
[3]孙宇辉,邓子新.聚酮化合物及其组合生物合成 [J].中国抗生素杂志,2006,1:6-14.
[4]胡又佳,朱春宝,朱宝泉.组合生物合成研究进展[J].中国抗生素杂志,2001,26,5:321-330.
[5]Donadio S,Staver M J,et al.Modular organization of genes required for complex polyketide biosynthesis[J].Science,1991,252:675.
[6]Cortes J,Wiesmann K E,et al.Repositioning of a domain in a modular polyketide synthase to promote specific chain cleavage.[J].Science,1995,268:1487.
[7]刘炳辉,曹元银,闫建芳,等.聚酮类化合物生物合成基因簇与药物筛选[J].生物技术通报,2008,4:30-33.
[8]罗师平,冷希岗.基于PCR的体外诱变技术 [J].国外医学生物医学分册,2005,28,3:188-192.
[9]周丽萍.聚合酶链式反应介导的定点突变方法探索[J].江苏大学学报(医学版),2003,13(4),364-365.
[10]DeBoer C,Meulman P A,et al.Geldanamycin,a new antibiotic[J].J.Antibiot,1970,23:442-447.
[11]Rascher A,Hu Z,et al.Cloning and characterization of a gene cluster for geldanamycin production in Streptomyces hygroscopicus NRRL 3602 [J].FEMS Microbiol.Lett,2003, 218:223-230.
[12]WoncheolKim,Dongho Lee,etal.Rational Biosynthetic Engineering for Optimization of Geldanamycin Analogues[J].ChemBioChem,2009,10:1243-1251.
[13]Treshchalin I D,Sletta H,et al. Comparative analysis of in vitro antifungal activity and in vivo acute toxicity ofthe nystatin analogue S44HP produced via genetic engineering[J].Antibiot.Khimioter,2005,50:18-22.
[14]Brautaset T,Sekurova O N,et al.Biosynthesis of the polyene antibiotic nystatin in Streptomyces noursei ATCC 11455:analysis of the gene cluster and deduction of the biosynthetic pathway[J].Chem.Biol,7:395-403.
[15]Espen Fjaervik,Sergey B Zotchev.Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei[J]. Appl Microbiol Biotechnol,2005,67:436-443.
[16]Trygve Brautaset,Ha vard Sletta,et al.Improved antifungal polyene macrolides via engineering of the nystatin biosynthetic genes in streptomyces noursei [J].Chemisry& Biology,2008,15:1198-1206.
Study of Analogues in Polyketides Compounds Type I Produced by Site Mutation
LIU Wei,GAO Ju-fang (College of Life and Environment Science,Shanghai Normal University,Shanghai 200234,China)
Site mutation is a method to change the DNA strand about the purpose strains by PCR(the change always towards the positive side),including add,delete orchange the bases.Changing the amino acid by homologous recombination via conjugation transfer between the wild type strain and the mutation plasmid,the function of protein is altered,even abolished.Then screening the right mutant and fermenting to find the new expected structural analogues.During the biosynthesis of polyketide natural products,there are many oxidationreduction reactions,so skip some step which the inactivation protein can not catalytic,get the change in the PKS skeleton and then the new analogues.
polyketide compounds;site mutation;structural analogues
1006-4184(2012)07-0025-05
2012-04-29
刘伟(1985-),男,微生物学专业硕士研究生。从事分子生物学相关研究。
指导老师:高菊芳(1962-),女,副教授。
猜你喜欢
肝博士(2022年3期)2022-06-30
科学导报(2022年28期)2022-05-24
食品安全导刊(2021年20期)2021-08-30
发明与创新·大科技(2019年6期)2019-09-06
天然产物研究与开发(2019年1期)2019-03-01
中国社区医师(2018年15期)2018-11-13
健康大视野(2018年16期)2018-11-05
科技创新与应用(2017年35期)2017-12-19
中国现代医生(2017年11期)2017-05-25
妇产与遗传(电子版)(2016年4期)2016-12-16
