
益母草颗粒工艺改进及质量标准提高的研究
2012-09-17 06:33郑艳春崔雅慧李俊芳董海荣
中国医药导报 2012年4期
郑艳春 李 君 崔雅慧 李俊芳 董海荣 朱 磊
承德颈复康药业集团有限公司,河北省中药新辅料工程技术研究中心,河北承德 067000
益母草颗粒现收载于《中国药典》2010年版一部,为妇科经产之要药,主要用于经闭、痛经、产后瘀血、腹痛等妇科疾病[1]。现在市面上规格是每袋装15 g,由于服用量相对较大,辅料用量多,不利于患者的服用,颗粒干燥时间长,没有控制质量的指标性成分。为此,通过对制剂工艺进行改造,制剂规格由原来的每袋装15 g变更为每袋装5 g,每袋疗效不改变,辅料量减少了60%以上,有效地控制了成品的质量,方便了患者的使用,增强患者依从性,降低企业生产成本。
1 仪器与试药
多功能提取罐(武汉制药机械厂),DBP-3 多功能制粒包衣机(重庆广厦生产),UV-2401 紫外-可见分光光度仪(日本岛津公司生产),医用型超声清洗器KQ-700DE(昆山市超声仪器有限公司),盐酸水苏碱对照品(中国药品生物制品检定所,供含量测定用,批号:110702-200709),硫氰酸铬铵(上海天莲精细化工有限公司,批号:2006-08-12-520),益母草药材、糊精、蔗糖均有承德颈复康药业集团提供,水为二次蒸馏水;其他试剂均为分析纯。
2 方法与结果
2.1 制剂工艺的研究
益母草颗粒原工艺为提取浸膏湿法挤压制粒,药物与辅料的比例相差较大,特别是干燥过程较慢,导致生产周期也较长,工艺改进后采用沸腾制粒技术制备颗粒,降低了制剂中辅料的用量,减少了颗粒干燥的时间,降低了生产的成本。
2.1.1 提取浸膏比例的选择
按照益母草提取工艺,提取浓缩后浸膏的比重为1.36~1.38,浸膏较黏稠,浓缩过程黏附在浓缩罐中的浸膏较多,损失较大,采用沸腾制粒技术,浸膏比重调整在1.15~1.30 范围内,浸膏损耗少,优选后发现浸膏在1.20~1.25 时,制粒效果较好。
2.1.2 药物、辅料比例的确定
根据试验设计规格,确定制剂中辅料的用量,以颗粒的制备状态、颗粒的粒度为考察点,分别设定糊精∶蔗糖比为5∶4、4∶5、3∶6 三个比例进行优选,而以糊精∶糖粉比例为 4∶5 时制得的颗粒大小均匀,粒度好,过程控制稳定。
2.1.3 工艺对比研究
试验中将变更前后两种工艺进行对比,结果采用喷雾制粒技术,浸膏相对密度降低到1.20~1.25;药物与辅料的比例降低到1.0∶2.2;干燥温度和干燥时间最为明显,由原来的22~24 h降低到2~3 h。结果制得的颗粒硬度适中,粒度均匀,明显缩短了生产周期,降低了生产成本。
2.2 质量研究
2.2.1 薄层色谱鉴别
取本品 5 g,研细,加乙醇 50 mL,超声 30 min,滤过,滤液蒸干,残渣加入0.1 mol/L盐酸溶液10 mL使溶解,滤过,滤液蒸干,残渣加乙醇5 mL使溶解,作为供试品溶液。取除去益母草,按益母草颗粒工艺制备的阴性样品,照供试品溶液的制备方法制成阴性样品溶液。另取盐酸水苏碱对照品,加乙醇制成每1 mL含2 mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,分别吸取供试品溶液、阴性样品溶液各10 μL及对照品溶液5 μL,点于同一硅胶G薄层板上,以乙酸乙酯-正丁醇-盐酸(3∶2∶1)为展开剂展开,取出,晾干,105℃烘干10 min,晾凉。喷以碘化铋钾-碘化钾碘(1∶1)试液,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。阴性样品无干扰。
2.2.2 方法学考察
2.2.2.1 线性关系考察 精密吸取盐酸水苏碱对照品溶液(1 mg/mL)4、6、8、10、12、14 mL, 分别置 25 mL 容量瓶中,加0.1 mol/mL的盐酸溶液使成20 mL,另取盐酸溶液20 mL,置另一25 mL容量瓶中,作为空白样品溶液,采用含量测定的方法测定,以盐酸水苏碱的浓度(X)为横坐标,吸收度的差值(Y)为纵坐标,绘制标准曲线,测得盐酸水苏碱的线性回归方程:Y=0.605 6X-0.003 1,r=0.999 4,线性范围为 0~8.153 6 mg。
2.2.2.2 精密度试验 精密称取上述对照品溶液6份,各8 mL,按照样品含量测定方法进行测定,测定的平均吸收度值为0.283 6,RSD 值为 0.26%(n=6)。
2.2.2.3 稳定性试验 取同一批号的供试品溶液,分别在放置0、2、4、6、8 h 后,测定其吸收度,结果样品溶液在 8 h 内稳定性良好,样品吸收度值的RSD值为1.34%(n=6),
2.2.2.4 重复性试验 取益母草颗粒(批号:080508)40 g,研细,精密称取 9份,分为三组,每组分别为 1.6、2.0、2.4 g,按“2.2.1”项下供试品溶液的制备方法制成供试品溶液,另取对照品溶液,按上述样品测定方法分别测定盐酸水苏碱的吸收度,记录吸收度值,计算样品含量。样品平均含量为18.7 mg/袋,RSD 值为 1.81%(n=9)。
2.2.2.5 加样回收率试验 取益母草颗粒(批号:080508)40 g,研细,精密称取9份,每份约1.0 g,精密加入不同量的盐酸水苏碱对照品溶液,按“2.2.1”项下供试品溶液的制备方法制成供试品溶液,按上述测定方法测定各样品的吸收度,计算回收率,盐酸水苏碱的平均回收率为98.5%(n=9),RSD=1.03%(n=9)。
2.2.2.6 专属性试验 按照处方配比,除去益母草,参照“2.2.1”项下供试品溶液的制备方法制成阴性样品溶液。按上述样品测定方法进行测定,阴性样品与空白试剂的吸收度无显著性差异,说明辅料不干扰盐酸水苏碱的测定。
2.2.2.7 对照品溶液的制备 精密称取105℃干燥至恒重的盐酸水苏碱对照品适量,用0.1 mol/L盐酸溶液溶解,制成每1 mL含盐酸水苏碱1 mg的溶液,即得。
2.2.2.8 供试品溶液的制备 取本品,研成细粉,精密称取2.0 g,置具塞锥形瓶中,精密加入乙醇50 mL,称定重量,超声处理30 min,放冷,再称定重量,用乙醇补足减失的重量,摇匀,滤过,精密量取续滤液25 mL,置蒸发皿中,水浴蒸干,精密加入0.1 mol/L盐酸溶液 10 mL,使溶解,加活性炭0.5 g,置水浴中加热半分钟,搅拌、滤过,滤液置25 mL量瓶中,用0.1 mol/L盐酸溶液分次洗涤蒸发皿及滤器,洗液并入同一量瓶中,备用。
2.2.2.9 测定方法 精密量取对照品溶液10 mL,置量瓶中,另取0.1 mol/L盐酸溶液20 mL,置25 mL量瓶中,作为空白样品溶液。在对照品溶液、0.1 mol/L盐酸空白样品溶液及上述备用供试品溶液的量瓶中,各精密加入新配制的2%硫氰酸铬铵溶液3 mL,摇匀,加0.1 mol/L盐酸溶液至刻度,摇匀,置冰浴中放置1 h,过滤,取续滤液,以0.1 mol/L盐酸溶液为空白,照紫外-可见分光光度法(《中国药典》2010年版一部附录ⅤA),在520 nm的波长处测定吸收度,用空白样品的吸收度分别减去对照品及供试品的吸收度,计算,即得。各批的测定结果见表1。
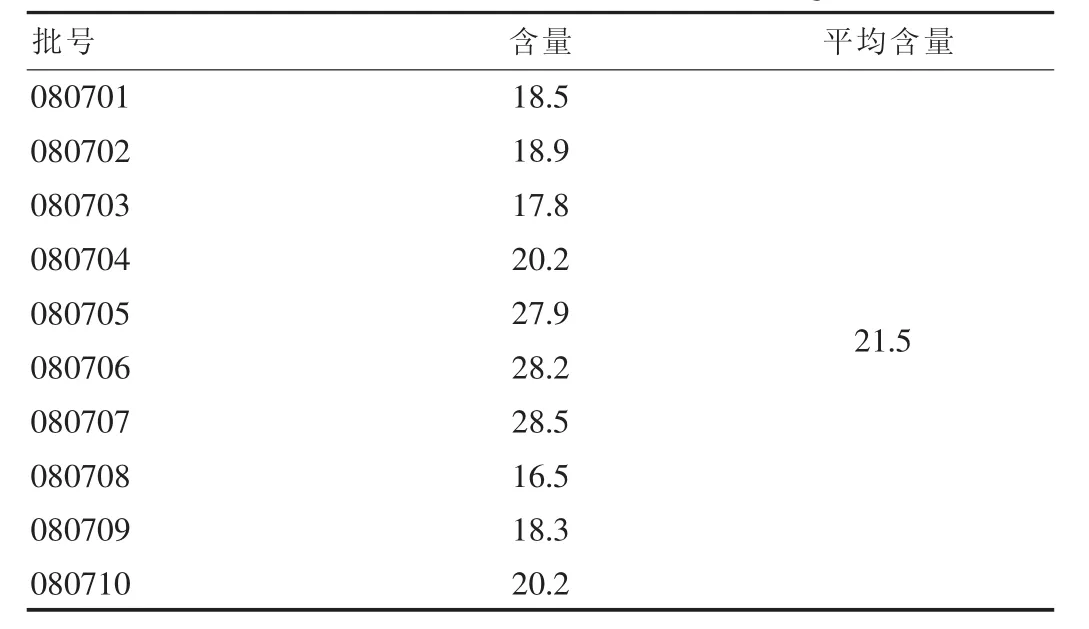
表1 益母草颗粒中盐酸水苏碱含量测定结果(mg/袋,n=3)
2.2.3 质量对比研究
取同一批次的药材,分别采用以上两种方法制备不同规格的益母草颗粒各三批,两种规格的样品所含的生药量相同,辅料的用量不同。从检测的结果看,鉴别项没有明显的变化,主斑点颜色清晰;含量测定项,平均含量分别为4.14 mg/g和4.18 mg/g,RSD=0.68%。提示沸腾制粒技术制备的颗粒不会对原规格颗粒的成分产生影响,但在一定的程度上提高了质量的可控性。
2.2.4 含量限度确定
从以上10 批样品测定结果看,益母草颗粒中盐酸水苏碱的含量在16.5~28.5 mg/袋范围内不等,平均含量为21.5 mg/袋。由于不同产地、规格、批次的益母草药材差异较大[2],导致制剂中盐酸水苏碱的含量也有较大的波动。因此,在上述测得平均值的基础上适当调整,根据生产的实际情况及检测的方法,将每袋颗粒含盐酸水苏碱确定为18.0 mg。
3 讨论
3.1 展开系统和条件的选择
对于益母草鉴别的展开系统,文献报道很多,常用的有正丁醇-盐酸-水(4.0∶1.0∶0.5)[3]、正丁醇-盐酸-乙酸乙酯(4.0∶1.5∶0.5)[3]、丙酮-无水乙醇-盐酸(10∶6∶1)[4],乙酸乙酯-正丁醇-盐酸(3∶2∶1)[5]等。 考察了各种展开剂对盐酸水苏碱的分离情况。前两种的展开系统,展开时间长,对薄层板的选择性强,展开的重现性差,斑点分散、不稳定;丙酮-无水乙醇-盐酸(10∶6∶1)展开系统展开速度快,样品的分离效果不明显;乙酸乙酯-正丁醇-盐酸(3∶2∶1)展开系统,用时短,分离度好,因此选用其作为该项的展开系统。
3.2 显色剂的选择
选用了盐酸水苏碱常用的显色剂:稀碘化铋钾试液、碘化铋钾试液、改良碘化铋钾试液、改良碘化铋钾-1%三氯化铁无水乙醇液(5∶1)等,均不能使斑点保持圆润、稳定不褪色,而使用碘化铋钾-碘化钾碘(1∶1)进行显色,盐酸水苏碱斑点显色快,清晰,稳定。
[1] 李文仕.益母草颗粒的质量控制[J].中国药师,2008,11(4):471-472.
[2] 张清民,朱颖虹,熊艳,等.益母草不同制剂盐酸水苏碱含量的测定[J].中成药,2001,23(6):437-438.
[3] 国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:1025-1026.
[4] 张玲,时延增,于宗渊,等.双波长薄层扫描法测定益母口服液中水苏碱的含量[J].中国药科大学学报,1996,27(l):15-16.
[5] 严优勺.益母草质量标准、提取工艺剂型的改进研究[D].广州:广州中医药大学,2005.
猜你喜欢
食品工业(2022年2期)2022-03-09
基层中医药(2021年9期)2021-06-05
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
中成药(2017年5期)2017-06-13
中成药(2017年5期)2017-06-13
中国药业(2014年12期)2014-06-06
中国药业(2014年24期)2014-05-26
中国药业(2014年21期)2014-05-26
中国药业(2014年4期)2014-05-09
