
以c-Met为靶点的小分子酪氨酸激酶抑制剂的研究进展
2012-09-12 06:02张喜全孟庆义
药学与临床研究 2012年6期
赵 锐,张喜全,孟庆义*
江苏正大天晴药业股份有限公司研究院,南京 210042
近年来分子生物学和肿瘤药理学研究表明,酪氨酸激酶(Protein Tyrosine Kinases,PTKs)作用的细胞信号转导通路在肿瘤的形成和发展中起着极其重要的作用。c-Met原癌基因属于PTKs家族中Ron亚族,其编码的c-Met蛋白是唯一肝细胞生长因子/离散因子(Hepatocyte Growth Factor/Scatter Factor,HGF/SF)的高亲和性受体。HGF/c-Met信号通路与血管新生和肿瘤生长过程密切相关,抑制该通路成为靶向抗肿瘤治疗的新手段。本文着重介绍近期已上市和处于临床后期的c-Met抑制剂的研究进展。
1 HGF/c-Met信号通路
c-Met蛋白是由50 kDa的细胞外α亚基和145 kDa的跨膜催化β亚基通过二硫键相连的异二聚体。c-Met包含有三个功能不同的结构域[1],见图1。HGF通过c-Met受体酪氨酸激酶催化域的Tyr1234和Tyr1235的磷酸化来激活c-Met激酶,进而与SH2(Src homology 2)结合,产生多种胞内信号转导[2]。尤其对一种细胞侵袭性生长起着重要作用。
正常HGF/c-Met信号通路涉及胚胎发育、组织损伤的修复等。正常细胞中c-Met RNA低水平表达,仅在组织损伤后短暂上升,随即又恢复正常水平。相反,肿瘤细胞中则存在HGF/c-Met的过度表达,例如人卵巢癌、鼻咽癌、子宫癌、胃癌、非小细胞肺癌、肾癌等癌症细胞中均观察到c-Met的高表达,且c-Met的过度表达和多种肿瘤的形成及预后密切相关,而将肿瘤细胞中异常活化的HGF/c-Met信号通路被阻断后,肿瘤细胞会出现细胞形态改变,增殖减缓,成瘤性降低,侵袭能力下降等一系列变化。

图1 c-Met结构示意图
2 c-Met抑制剂
目前,以HGF/c-Met为靶点的肿瘤分子靶向治疗药物主要包括有HGF/c-Met拮抗剂、小分子c-Met抑制剂、c-Met反义核酸分子和c-Met抗体等。对配体与c-Met激酶复合物晶体结构及小分子c-Met抑制剂构效关系的研究显示:小分子c-Met抑制剂是对c-Met的激酶催化结构域进行竞争性结合而抑制其作用。c-Met的激酶催化结构域为一高度保守的催化核心区域,是通过亚基折叠而成的铰链(Hinge Region)连接的双叶结构(如图2显示),激酶含有一个起始端为DFG(Asp-Phe-Gly)序列,终止端为APE(Ala-Pro-Glu)序列的环状活化区域,其中DFG序列中的Asp和Phe朝向ATP结合位点时,记为DFG-in,为激酶活化构象,当DFG序列中的Phe朝向ATP结合位点时,记为DFG-out,为激酶的非活化构象。针对DFG-in活化构象的抑制剂为TypeⅠ抑制剂,由于TypeⅠ抑制剂要竞争性抑制激酶的铰链区域,而铰链区域的结构较为固定,因此TypeⅠ抑制剂的可改动空间较小。DFG-out非活化构象与DFG-in构象相比,多出一个额外的疏水区域,也称变构位点。变构位点的氨基酸序列不如铰链结构的氨基酸序列保守,以此为基础,可设计具有更多选择性的小分子c-Met抑制剂。

图2 配体与c-Met激酶复合物晶体结构
化学合成的小分子c-Met抑制剂研究进展较快,已有不少药物分子上市或进入临床后期,例如Crizotinb、Foretinib、Cabozantinib、Tivantinib 和 Amuvatinib等,现简述如下:
2.1 Crizotinib
Crizotinib(克里唑替尼,PE2341066,商品名:XalkoriTM,图3)是美国辉瑞公司研发的HGF/c-Met和间变型淋巴瘤激酶(anaplastic lymphoma kinase,ALK)双重作用的酪氨酸激酶抑制剂,已于2011年8月被FDA批准上市,用于治疗ALK阳性的局部晚期或转移的非小细胞肺癌(NSCLC)。该药用于治疗实体瘤和恶性淋巴瘤的研究处于临床Ⅰ/Ⅱ期阶段[3]。
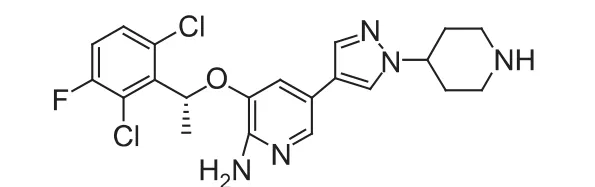
图3 克里唑替尼
Crizotinb制剂规格为250 mg/粒和200 mg/粒,口服一日2次。
在167名受试者的药代动力学实验结果显示,Crizotinib的血药浓度达峰时间(tmax)中位数为4 h,平均表观终末半衰期(t1/2)为42 h。本品多次给药表现出非线性的药动学特征,连续给药15天和28天的表观清除率(CL/F)的几何平均值分别为64.5 L·h-1和 60.1 L·h-1,均低于单次给药后的 CL/F(100 L·h-1)。
根据美国临床肿瘤学会2011年公布的研究结果表明,已进行的多中心单臂临床Ⅰ/Ⅱ期试验研究,以RECIST的客观缓解率(ORR)和缓解持续时间(DR)为评估标准,在 PROFILE1005中,ORR为50%,包括1例完全缓解和67例部分缓解,中位治疗时间为22周,而治疗的前8周,获得了79%的ORR,其中位缓解持续时间为41.9周;在扩展试验1001中,ORR为61%,包括2例完全缓解和69例部分缓解,中位治疗时间为32周,治疗前8周,获得55%ORR,中位缓解持续时间为48.1周,接受Crizotinib的ALK阳性NSCLC患者获得明显的幻觉,生存率有显著的提高。Crizotinib的临床试验中常见的副作用有视力障碍、恶心、腹泻、便秘等。
2.2 Foretinib
Foretinib(XL880,GSK1363089,图 4)是法国Exelixis公司研发的作用c-Met和血管表皮生长因子受体-2(VEGFR-2)的双重酪氨酸激酶抑制剂,后续研发权利已转让给葛兰素史克公司,目前该药针对乳腺癌、头颈部癌、肾癌、胃癌的研究处于临床Ⅱ期阶段,针对肺癌和肝癌的研究处于临床Ⅰ期阶段[4]。
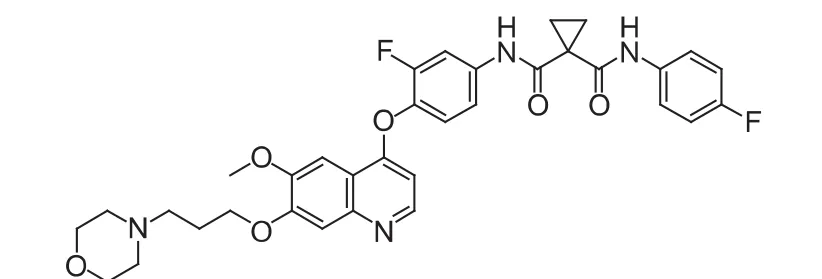
图4 XL880
体外试验[5]表明,Foretinib对于c-Met和VEGFR有强效抑制作用,其IC50分别为0.4 nmol·L-1和0.9 nmol·L-1,而抑制 HGF和 VEGF诱导的HMVEC-L肿瘤细胞转移的IC50分别为17 nmol·L-1和5 nmol·L-1。在裸鼠接种B16F10实体瘤细胞的试验中,Foretinib显示出剂量依赖的抑制作用,在30 mg·kg-1和 100 mg·kg-1组中,Foretinib 分别使肿瘤体积缩小了64%和87%。
在已进行的Ⅰ/Ⅱ期临床试验中[6],给予38名患者每天一次30mg的Foretinib进行疗效评估,试验数据显示客观响应率(ORR)为24%,病情稳定率(DSR)为79%,中位进展期为 4.2个月,结果表明Foretinib为有效且易于耐受的抗肿瘤药物。在74名患者参与的Ⅱ期临床试验中,每天给予240 mg Foretinib,总响应率至少为13.5%,无进展生存期为9.3个月,1年生存率为70%,同时在68名得到充分评估的患者中,有50%的患者肿瘤直径有2~75%的减小。Foretinib常见的副反应主要有高血压(36%)、食欲减退(23%)和发烧(21%)。
2.3 Cabozantinib
Cabozantinib(XL184,图 5)是 Exelixis原研药物,后与百时施贵宝联合开发的双重c-Met和VEGFR-2的抑制剂,目前该药治疗甲状腺髓样癌的研究处于临床Ⅲ期阶段[7]。
在已进行的体内体外实验中,Cabozantinib均表现出对c-Met和VEGFR2磷酸化的强效抑制作用。在裸鼠模型中,Cabozantinib可以控制肿瘤细胞的转移途径,从而达到减小内皮细胞的增殖和肿瘤细胞的生长[8]。
已完成的398例进展性可测量癌症患者的RDT的Ⅱ期临床试验[9]结果显示,每日给药剂量为100 mg。Cabozantinib对多种晚期肿瘤具有较高的疾病控制率,并能缩小甚至消除骨转移病灶,单药治疗12周后,肝癌、前列腺癌和卵巢癌的疾病稳定率分别达到76%、71%和58%,而对黑色素瘤、乳腺癌和非小细胞肺癌的疾病控制率分别为45%、45%和40%,其中68例骨转移患者中,有59例骨转移消失或消除,包括乳腺癌、前列腺癌和黑色素瘤患者,骨扫描显示转移病灶部分或完全消失,所有患者在治疗6周后症状得到明显改善。
2.4 Tivantinib
Tivantinib(ARQ197,图 6)是美国 ArQule 公司与日本第一三共制药等公司联合研发的小分子c-Met抑制剂,目前该药治疗非小细胞肺癌的研究处于临床Ⅲ期阶段,针对肝癌、胰腺癌、胃癌等处于临床Ⅱ期阶段。
Tivantinib不仅抑制细胞增殖,还抑制c-Met过度表达的肿瘤细胞中半胱氨酸激酶依赖的细胞凋亡。Tivantinib可明显抑制裸鼠上接种的结肠癌细胞HT-29,胃癌细胞MKN-45,乳腺癌细胞MDA-MB-231的肿瘤生长[10]。
在TivantinibⅡ期临床的随机安慰剂对照双盲试验[11]中,107名不可手术的肝癌或对于其他一线化疗药物无效或不可耐受的患者中,分别给予每天2 次 360 mg,240 mg或安慰剂,以进展时间(TTP)为终点的ITT统计显示,相较于安慰剂组,Tivantinib组有58%的提高,其中中位总生存期Tivantinib组为7.2个月,而安慰剂组只有3.8个月;中位进展时间(Median TTP)Tivantinib组为2.9个月,而安慰剂组为1.5个月;中位无进展生存期(Median PFS)Tivantinib组为2.4个月,而安慰剂组为1.5个月。同时对于高Met表达的肿瘤患者,Tivantinib疗效较为明显。上述临床试验结果证明Tivantinib作为单独使用的药物,对于不可手术的肝癌患者显示较好的疗效。
Tivantinib常见的副反应主要有疲劳、恶心、呕吐等,罕见不良反应有转氨酶升高,肝酶升高。
2.5 Amuvatinib
Amuvatinib(MP-470,图 7)是英国 Astex 公司研发的多靶点酪氨酸激酶抑制剂,主要作用于c-Met、c-Ret、c-Kit、PDGFR 和 FLT3 等多个靶点,目前该药治疗小细胞肺癌(SCLC)的研究处于临床Ⅱ期阶段,治疗实体瘤的研究处于临床Ⅰ期阶段。
Amuvatinib可抑制肺癌细胞中H1299的RAD51蛋白的表达和同源重组[12],进而可减少肿瘤细胞的抗药性。在100个病人参与的Amuvatinib与其他5种标准化疗方案联合用药的Ib临床实验[13]显示,Amuvatinib与紫杉醇,Amuvatinib与依托泊苷合用组显示出较强的抗肿瘤活性,同时小细胞肺癌和神经内分泌肿瘤对于Amuvatinib较敏感,46%患者的肿瘤得到了部分控制。
2.6 其他
除了前述的药物外,还有不少候选药物如百时施贵宝公司的BMS-777607(图8),Sugen公司 SU-11274(图 9)和辉瑞公司 PHA-665752(图 10)等已进入临床前期阶段,展现出良好的前景。

图5 XL184
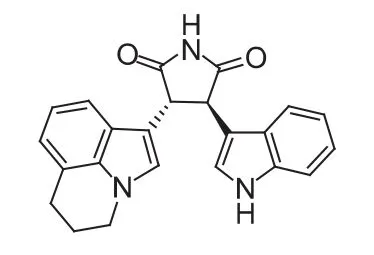
图6ARQ197
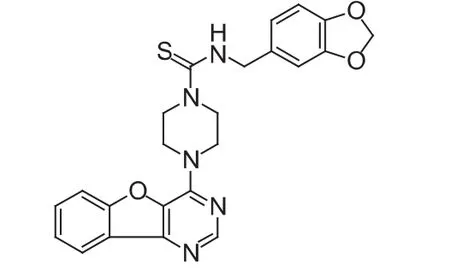
图 7 MP-470
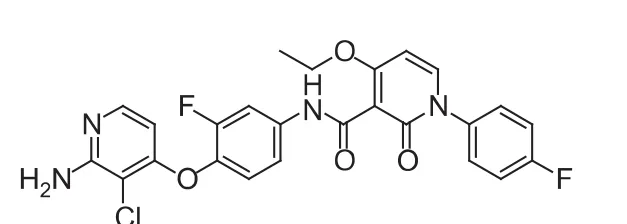
图 8 BMS-777607
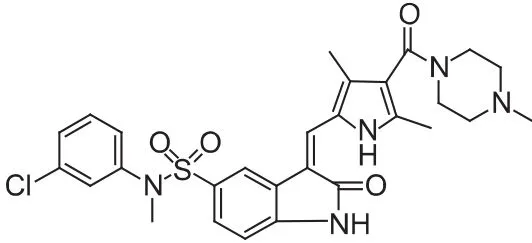
图 9 SU-11274

图 10 PHA-665752
3 结 语
综上所述,小分子c-Met酪氨酸激酶抑制剂通过作用于c-Met受体,抑制c-Met自身磷酸化位点的磷酸化,阻止酪氨酸激酶的激活,进而抑制其下游细胞信号的转导,尤其是PI3K及Ras途径的信号转导,抑制肿瘤细胞的产生和转移,同时抑制肿瘤血管新生等作用,而起到抗肿瘤的目的。c-Met抑制剂相较于传统的细胞毒类抗肿瘤治疗药物来说,具有作用机制清楚,疗效强大,毒副作用低等优点,其研究开发前景广阔。随着HGF/c-Met信号转导通路及其与肿瘤作用机制研究的深入,会有更多的小分子c-Met酪氨酸激酶抑制剂被开发出来,将对攻克肿瘤、改善人类健康起到重要作用。
[1]Weon-Kyoo Y,Donald MM.The hepatocyte growth factor/c-Met signaling pathway as a therapeutic target to inhibit angiogenesis[J].BMB Reports,2008,41(12)∶833-9.
[2]Comoglio PM.Pathway specificity for Met signaling[J].Nat Cell Biol,2001,3(7)∶E161-2.
[3]抗肿瘤药 Crizotinib[J]. 药学进展,2012,36(1):42-3.
[4]Seiwert T,Swann S,Kurz H,et al.A phase II study of the efficacy and safety of Foretinib∶a novel receptor tyrosine kinase inhibitor,given on an intermittent 5-days-on/9-days-off (5/9)schedule in patients with recurrent or metastatic squamous cell cancer of the head and neck[J].Mol Cancer Ther,2009,8(12Suppl)B6.
[5]Qian F,Engst S,Yamaguchi K,et al.Inhibition of tumor cell growth,invasion,and metastasis by EXEL-2880(XL880,GSK 1363089),a novel inhibitor of HGF and VEGF receptor tyrosine kinases[J].Cancer Res,2009,69(20)∶8009-16.
[6]Jhawer M,Kindler HL,Wainberg Z,et al.Assessment of two dosing schedules of GSK1363089 (GSK089),a dual MET/VEGFR2 inhibitor,in metastatic gastric cancer(GC)∶Interim results of a multicenter phase II study[J].J Clin Oncol,2009,27(15,Suppl)∶Abst4502.
[7]Salgia R,Sherman S,Hong DS,et al. A phase I study of XL184,a RET,VEGFR2 and Met kinase inhibitor,in patients with advanced malignancies,including patients with medullary thyroid cancer(MTC)[J].J Clin Oncol,2008,26(15,Suppl)∶Abst3522.
[8]Yakes FM,Chen J,Tan J,et al.Cabozantinib(XL184),a novel MET and VEGFR2 inhibitor,simultaneously suppresses metastasis,angiogenesis,and tumor growth[J].Mol Cancer Ther,2011,10(12)∶2298-308.
[9]Nechushtan H,Edelman G,Jersalem G,et al.Phase 2 results of XL184 in a cohort of patients with advanced melanoma[C].22nd EORTC-NCI-AACR Symp Mol Targets Cancer Ther,2010,Abst398.
[10]Munshi N,Jeay S,Li Y,et al.ARQ 197,a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity[J].Mol Cancer Ther,2010,9(6)∶1544-53.
[11]Sandler A,Schiller JH,Hirsh V,et al.A phase III randomized,double-blind,placebo-controlled study of erlotinib plus ARQ197 versus erlotinib plus placebo in previously treated subjects with locally advanced or metastatic,nonsquamous,non-small cell lung cancer(NSCLC)[C].47th Annu Meet Am Soc Clin Oncol(ASCO),2011,Abst TPS217.
[12]Zhao H,Luoto KR,Meng AX,et al.The receptor tyrosine kinase inhibitor amuvatinib (MP470)sensitizes tumor cells to radio-and chemo-therapies in part by inhibiting homologous recombination[J].Radiother Oncol,2011,101(1)∶59-65.
[13]Tolcher AW,Mita M,Gordon M,et al.Clinical responses in highly refractory solid tumor patients with oral MP-470,a multi-targeted tyrosine kinase inhibitor,in combination with standard of care chemotherapy regimens∶Preliminary report from a multi-institutional phase-1b clinical trial[J].Eur J Cancer Suppl,2008,6(12)∶Abst 403.
猜你喜欢
保健与生活(2022年5期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
皮肤病与性病(2021年3期)2021-07-30
中外文摘(2020年13期)2020-08-01
福建基础教育研究(2020年4期)2020-05-28
时代英语·高三(2019年4期)2019-09-03
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
恋爱婚姻家庭·青春(2016年10期)2016-10-10
中学生理科应试(2014年12期)2015-01-15
