
PS@Au核壳结构纳米催化剂的制备及性能研究
2012-09-04 11:41刘大博祁洪飞
材料工程 2012年7期
刘大博,祁洪飞,成 波
(北京航空材料研究院,北京100095)
PS@Au核壳结构纳米催化剂的制备及性能研究
刘大博,祁洪飞,成 波
(北京航空材料研究院,北京100095)
采用化学镀工艺,制备了催化性能优良的PS@Au纳米催化剂,负载于表面的Au颗粒粒径约为16nm,结构致密,单分散程度较高。利用扫描电子显微镜(SEM)和紫外-可见分光光度计(UV-Vis),结合催化性能测试,研究了原料加入次序对PS@Au纳米核壳结构催化性能的影响。结果表明:PS@Au纳米核壳结构较大的比表面积是其显著提高亚甲基蓝发色团的降解率以及具有较高催化效果的根本原因,较高的纳米Au负载率和颗粒粗糙度有助于其催化性能的提高。关键词:纳米催化剂;核壳结构;聚苯乙烯微球;催化性能
核壳型贵金属纳米粒子是近年来备受关注的一类新型功能材料,其通常以Au,Ag,Pt,Pd等贵金属作为壳层或核层,通过核壳尺寸设计和化学成分调制,可赋予其新奇的物理和化学性质。作为催化剂,核壳型贵金属纳米结构具有单分散性能好、颗粒稳定性高、比表面积大、催化活性高、易于回收以及使用寿命长等诸多优势[1]。研究较多的核壳结构主要包括非金属@贵金属、贵金属@非金属、金属@贵金属和贵金属@贵金属等结构[2-5],其在材料科学、生物物理、分子电子学以及基于表面增强效应的荧光工程学领域具有极其广泛的应用前景[6,7]。尤其随着各类航空飞行器和空间攻防技术新概念的提出和逐步实现,燃料电池在航空发动机、火箭、军用飞机以及通讯指挥系统等航空领域的应用潜力大幅提升[8,9]。扮演着电化学反应“工厂”作用的贵金属纳米粒子作为燃料电池的核心材料更是备受各国军方的重视,已然成为航空制造领域的研究热点之一[10]。
由于聚苯乙烯(PS)微球为高分子材料,可利用化学表面改性,在其表面生成功能性官能团形成催化活性中心,通过化学反应生长贵金属纳米颗粒层。因此,PS@Au核壳结构可兼具有机相的易加工性和韧性,并且结构稳定,克服了其他核壳结构中不易避免的晶格失配和负载质量差等缺陷[11]。目前,制备粒径连续可控的高单分散性PS微球(粒径小于500nm,分散系数≤0.05)以及包覆均匀的贵金属纳米粒子仍然是该领域的技术难点。
本工作采用乳液聚合技术,通过对聚合反应条件的研究,制备了粒径120~350nm的单分散PS微球。继而采用化学镀工艺,重点研究原料加入顺序对Au纳米层质量的影响,制备了催化性能优良的PS@Au核壳结构纳米催化剂。
1 实验方法
1.1 试剂
PS纳米微球:过硫酸钾(KPS)、正丁醇(C4H9OH)、十二烷基硫酸钠(SDS)均为分析纯,苯乙烯(C8H8)为化学纯。除苯乙烯外,所有试剂均不做进一步纯化处理。
化学镀Au:无水乙醇、碳酸钠、磷酸钠、重铬酸钾、浓硫酸、氯化亚锡、氯化钯、次亚磷酸钠、氢氧化钠、氯金酸、亚硫酸钠、硫代硫酸钠、柠檬酸钠、酒石酸、氯化铵,均为分析纯。
实验用水均为去离子水。
1.2 PS纳米微球的制备
将一定量苯乙烯倒入分液漏斗中,依次用0.1mol/L NaOH溶液和去离子水洗涤以除去阻聚剂,再用无水硫酸钠干燥除水,最后减压蒸馏得到纯化的苯乙烯,4℃下保存待用。将0.12g SDS溶解于90m L去离子水,导入三口烧瓶,通氮气搅拌10min,控制搅拌速率为300r/min。升温至80℃后,加入0.15g KPS溶液。5min后滴加入5g苯乙烯与0.10g正丁醇的混合物,滴加时间为30min。80℃聚合反应3h,最后冰浴冷却至室温,得到PS纳米微球。
1.3 PS微球预处理
预处理目的是在微球表面形成活化面以及具有活性的自催化中心,其工艺主要包括粗化、敏化和活化处理。采用30g/L重铬酸钾和60m L/L的浓硫酸混合液作为粗化剂对微球进行粗化处理,在40℃下将微球放入混合液中处理1.5h,随后分别用10%氢氧化钠溶液和去离子水进行洗涤2遍,其目的是在微球表面形成细微的凹坑以增加其表面能,提高表面活性。采用一步法进行敏化活化处理,首先用去离子水稀释氯化钠、氯化亚锡和氯化钯,然后在搅拌下进行混合。混合液在40℃和50℃下分别放置1h。然后对微球表面进行敏化活化处理,处理温度为室温,时间为1h。最后用去离子水洗涤,得到预处理的微球。其目的是在微球表面均匀的吸附一层Sn2+,利用其还原性将Pd2+还原为Pd,形成活性中心,使贵金属粒子易吸附于PS微球表面。
1.4 化学镀Au
将20g柠檬酸钠、1g酒石酸和5g氯化铵,溶于100m L去离子水中,加入5g氯金酸、12g亚硫酸钠和5g硫代硫酸钠,混合均匀。随后加入超声分散的PS微球溶液,搅拌30min后对溶液过滤分离,用水和乙醇对其反复洗涤过滤,随后在40℃下真空干燥,得到PS@Au纳米催化剂。本工作采用二氧化硫还原法作为镀金方法,其化学反应式为:

1.5 表征
样品的微观形貌由FEI-SIRION型扫描电子显微镜(SEM)观测。用显微镜照相法拍摄微球样品的照片,随机抽取100个微球,测其直径并进行统计处理。PS微球的直径通过SEM进行测量和标定。微球的平均粒径、标准偏差和分散系数按下列公式计算:

式中:di为单个微球的直径;d为微球平均粒径;n为样本容量;δ为标准偏差;ε为分散系数。
吸光度曲线由U-3010型紫外可见分光光度计(UV-Vis)测定。
2 结果与讨论
2.1 SDS浓度对微球的影响
研究了乳化剂(SDS)浓度对微球粒径及其分布的影响,结果如图1所示。可见,随着乳化剂用量的增加,微球粒径逐渐减小,其粒径分布则呈现出不规则变化但不明显,微球分散系数始终保持在0.035以下,所制备微球具有较高的单分散性。结果表明,增加SDS的用量可在不降低单分散性的情况下合成小粒径PS微球。随着乳化剂用量越多,体系中形成的胶束就越多,产生的乳胶粒就更多;另外较高的乳化剂用量,可在聚合期间充足补充乳胶粒的增长,最终形成更多的乳胶粒。因此,在保持单体量不变的情况下,微球粒径随着乳化剂浓度的增加而减小。

图1 SDS用量对微球粒径及粒径分布的影响Fig.1 Effect of dosage of SDS on diameter and particle size distribution of PS particles
通过对乳化剂的调制,最终合成了粒径120~350nm连续变化的单分散PS微球,典型结果如图2所示。可见,本工作制备的纳米量级微球球形度较高,粒径均一,单分散性能较高。

图2 不同粒径的聚苯乙烯微球扫描电镜照片 (a)120nm;(b)160nm;(c)300nm;(d)350nmFig.2 SEM images of monodisperse PS microspheres with different diameters (a)120nm;(b)160nm;(c)300nm;(d)350nm
图3为所制备PS微球的DSC曲线,可见,PS微球在103.2℃附近出现了一个明显的吸热峰,由此可知其玻璃化温度Tg=103.2℃,结果表明所制备微球完全可承受预处理和化学镀Au过程中的热作用。
2.2 原料加入次序对镀层质量的影响
由于氯金酸的酸性较强,需在金源液中加入稳定剂后才能加入改性PS微球。另外,Au离子可与稳定剂中的柠檬酸钠发生反应,因此需在稳定剂之前加入络合剂,络合Au离子使其成为稳定的金源。最后,还原剂亚硫酸钠和络合剂硫代硫酸钠的加入次序对PS@Au核壳材料的表面形貌有一定影响。图4(a)为先加还原剂得到的样品形貌,图4(b)为先加络合剂样品的表面形貌。

图3 聚苯乙烯微球的DSC曲线Fig.3 DSC performance of PS microspheres

图4 不同原料加入次序制备的PS@Au纳米催化剂的扫描电镜照片 (a)先加还原剂;(b)先加络合剂Fig.4 SEM images of PS@Au nano-catalyst prepared with different adding order(a)reductant added first;(b)complexing agent added first
对比图4(a)与图4(b)可以发现,还原剂和络合剂的加入次序不同,导致所得样品的表面形貌有很大差异,先加入亚硫酸钠的样品表面Au层颗粒较小,而先加入硫代硫酸钠的样品则颗粒粗大,表面粗糙。表1为二者的EDX数据,可见不同的原料加入次序会导致颗粒表面Au负载量不同。先加入亚硫酸钠样品的Au原子分数为22.12%,而先加入硫代硫酸钠样品的Au原子分数可达到41.40%。结果表明,在本实验条件下,先加入硫代硫酸钠会增大镀层表面Au层的粗糙度及Au的负载量,这显然有助于提高PS@Au核壳纳米材料的催化性能。

表1 不同原料加入次序制备的PS@Au纳米催化剂表面的EDX数据Table 1 EDX data of PS@Au nano-catalyst prepared with different adding order
2.3 纳米核壳结构的催化性能
通过工艺优化,采用氯金酸→硫代硫酸钠→亚硫酸钠→稳定剂→PS的原料加入次序,在粒径300nm的PS微球表包覆了Au纳米颗粒,结果如图5所示。

图5 PS@Au纳米催化剂的SEM照片 (a)粒径300nm催化剂形貌;(b)局部放大Fig.5 SEM images of PS@Au particles (a)morphology of catalyst in 300nm;(b)magnification of(a)
可见,本工作制备的PS@Au核壳结构Au镀层结构致密,纳米粒子粒径均一,单分散程度较高。经SEM测量,Au纳米粒子粒径约为16nm。表2给出了其EDX数据,可见,PS@Au核壳结构表面的Au原子分数达到50.49%,质量比达到93.02%,Au纳米粒子成功包覆在PS微球表面。

表2 粒径300 nm的PS@Au核壳结构表面的EDX数据Table 2 EDX data of PS@Au nano-catalyst in 300nm
测试了PS@Au纳米核壳结构的催化性能,反应液由10m L亚甲基蓝溶液(50mg/L)和10m L硼氢化钠溶液(2.7g/L)配制而成。取三个反应液样品,其中两个样品中分别加入先加络合剂和先加还原剂制备的PS@Au纳米核壳结构,并取第三个样品作为空白样对比。保持容器静止,反应1h后通过测量剩余溶液的吸光度评价其催化性能,结果如图6所示。由于亚甲基蓝(MB)在可见光波段664nm处有明显的吸收峰,硼氢化钠与亚甲基蓝的生色团发生加氢反应后,使该处峰值降低。由图6可见,放入PS@Au纳米核壳结构的两个反应液样品在664nm处的吸收峰值有明显下降,分别为0.2408和0.2113,远低于放入空白样品的0.3594,PS@Au纳米核壳结构显著提高了亚甲基兰发色团的降解率,表现出优良的催化效果。本研究制备的核壳结构纳米催化剂Au颗粒负载均匀致密,单分散程度较高,粒径仅在16nm左右,具有较大的比表面积,这是其取得优良催化性能的根本原因。另外,先加入络合剂制备的核壳结构的催化性能最高,得益于其表面较高的Au负载量和颗粒粗糙度。
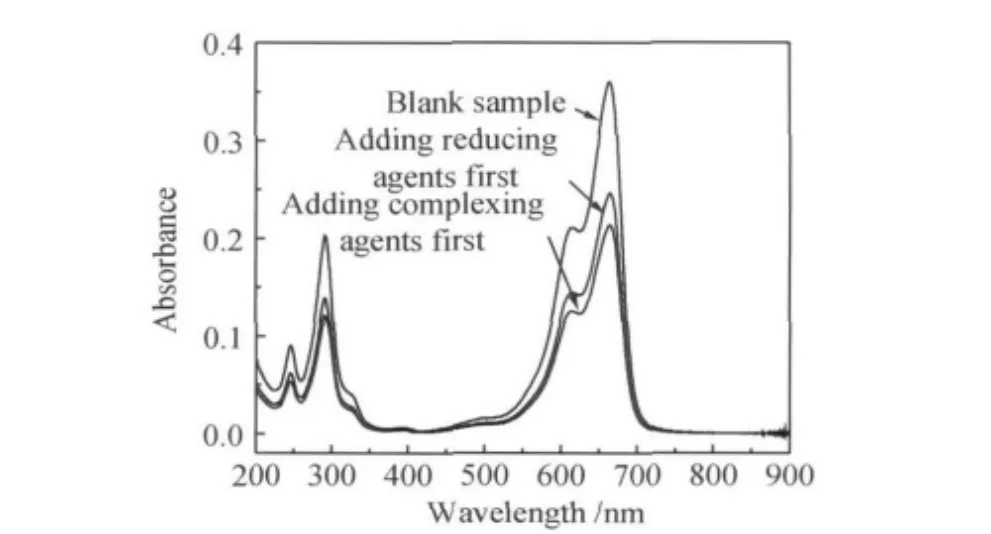
图6 催化降解亚甲基蓝的UV-Vis吸收谱Fig.6 UV-Vis absorption spectra of catalytic degradation of MB
3 结论
(1)采用乳液聚合法,通过对SDS的调制,制备了粒径120~350nm连续变化的PS微球,其分散系数均在0.035以下,表现出优良的单分散性。
(2)原料加入次序对PS@Au纳米核壳结构的催化性能有显著影响,较高的纳米Au负载率和颗粒粗糙度有助于其催化性能的提高。
(3)通过工艺优化,采用氯金酸→硫代硫酸钠→亚硫酸钠→稳定剂→PS的原料加入次序,制备了PS@Au纳米催化剂,Au纳米颗粒层结构致密,粒子粒径均一,单分散程度较高,粒径约为16nm。PS@Au纳米核壳结构较大的比表面积是其显著提高亚甲基蓝发色团的降解率以及具有较高催化效果的根本原因。
[1] PRODAN E,RADLOFF C,HALAS N J,et al.A hybridization model for the plasmon response of complex nanostructures[J].Science,2003,302(5644):419-422.
[2] SARDAR R,FUNSTON A M,MULVANEY P,et al.Gold nanoparticles:past,present,and future[J].Langmuir,2009,25(24):13840-13851.
[3] CHO S J,SHAHIN A M,LONG G J,et al.Magnetic and mössbauer spectral study of core/shell structured Fe/Au nanoparticles[J].Chemistry of Materials,2006,18(4):960-967.
[4] AZZAM T,EISENBERG A.Monolayer-protected gold nanoparticles by the self-assembly of micellar poly(ethylene oxide)-b-poly(ε-caprolactone)block copolymer[J].Langmuir,2007,23(4):2126-2132.
[5] FENG X,MAO C,YANG G,et al.Polyaniline/Au composite hollow spheres:synthesis,characterization,and application to the detection of dopamine[J].Langmuir,2006,22(9):4384-4389.
[6] PARK H Y,SCHADT M J,WANG L,et al.Fabrication of magnetic core@shell Fe oxide@Au nanoparticles for interfacial bioactivity and bio-separation[J].Langmuir,2007,23(17):9050-9056.
[7] 杨晓峰,董相廷,周艳慧,等.贵金属核壳纳米粒子最新研究进展[J].稀有金属材料与工程,2009,38(2):368-372.
[8] 李国超,简弃非,孙绍云.质子交换膜燃料电池在军事重的应用前景[J].兵工学报,2007,28(4):487-490.
[9] 王东,张伟,刘向.质子交换膜燃料电池及其空间应用[J].上海航天,2005,(2):39-42.
[10] 刘宾,廖世军,梁振兴.核壳结构:燃料电池中实现低铂电催化剂的最佳途径[J].化学进展,2011,23(5):852-859.
[11] CORBIERRE M K,CAMERON N S,SUTTON M,et al.Gold nanoparticle/polymer nanocomposites:dispersion of nanoparticles as a function of capping agent molecular weight and grafting density[J].Langmuir,2005,21(13):6063-6072.
Preparation of PS@Au Nano-catalyst and Its Catalytic Performance
LIU Da-bo,QI Hong-fei,CHENG Bo
(Beijing Institute of Aeronautical Materials,Beijing 100095,China)
PS@Au nano-catalyst with excellent catalytic performance was prepared by electroless plating technology,and the diameters of Au nanoparticles were around 16 nm with a narrow distribution.The influence of adding order for raw materials on the catalytic performance was investigated by SEM,UV-Vis spectrometer,and the detection of catalytic performance.It was found that larger surface area was responsible for the improvement of degradation rate of MB chromophore and the excellent catalytic performance of PS@Au nano-catalyst.An increase in the loading and surface roughness of Au nanoparticles could help to promote the catalytic performance of the PS@Au nano-catalyst.
nano-catalyst;core-shell structure;PS microspheres;catalytic performance
TQ426.6
A
1001-4381(2012)07-0001-04
2011-12-30;
2012-05-03
刘大博(1969—),男,硕士,高级工程师,主要从事贵金属功能材料领域的研究,联系地址:北京81信箱72分箱(100095),E-mail:liudabo600@yahoo.com.cn
