
DNA聚合酶Ⅲ全酶的功能和结构的发现
2012-08-21 10:14向义和
自然杂志 2012年4期
向义和
教授,清华大学物理系,北京100084
DNA聚合酶Ⅲ全酶的功能和结构的发现
向义和
教授,清华大学物理系,北京100084
DNA聚合酶Ⅲ DNA聚合酶Ⅲ*共聚物酶Ⅲ*α核心聚合酶 ε亚基 β亚基滑动夹子 γ复合物
介绍了DNA聚合酶Ⅲ和聚合酶Ⅲ*的发现,DNA聚合酶Ⅲ全酶形式的提出以及全酶亚基的分离。同时,介绍了DNA聚合酶Ⅲ全酶的结构和功能,其中包括α核心的聚合酶功能,ε亚基的3′→5′外切核酸酶活性,β亚基滑动夹子的功能以及γ复合物的结构与功能。
DNA聚合酶Ⅲ全酶(DNA polymeraseⅢholoenzyme)是DNA复制中最主要的酶,是能够进行引物链的延伸并完成DNA的前导链和后随链合成的酶。全酶是一个多亚基酶。它具有很快的复制速率和很高的延伸能力,大约每秒合成750个核苷酸,与在大肠杆菌中观察到的复制叉运动速率是一致的,比DNA聚合酶Ⅰ(polⅠ)的每秒合成10~20个核苷酸的速率快。这样快的速率来自于全酶的高度持续合成能力。
笔者分4部分介绍了DNA聚合酶Ⅲ全酶的结构和功能发现的历程。第1部分介绍了DNA聚合酶Ⅲ(polⅢ)的发现。第2部分介绍了DNA聚合酶Ⅲ*的发现和全酶形式的分离。第3部分介绍了DNA聚合酶Ⅲ全酶亚基的分离。第4部分介绍了DNA聚合酶Ⅲ全酶的结构和功能,其中包括核心α的聚合酶功能,ε亚基的3′→5′外切核酸酶活性,β亚基滑动夹子的功能以及γ复合物的结构与功能。
1 DNA聚合酶Ⅲ的发现
在DNA聚合酶Ⅰ被分离出来后不久,大量实验事实证明它不适于庞大的DNA复制过程。第一,它添加核苷酸的速度(600个/min)仅为大肠杆菌细胞中复制叉移动速度的1/100左右。第二,DNA聚合酶Ⅰ的连续合成的能力相对较低。第三,对基因的研究表明,许多基因和蛋白质参与了复制过程,显然DNA聚合酶Ⅰ不会单独起作用。第四,约翰·凯恩斯(John Cairns)分离出一个因基因突变而不能合成有活性的DNA聚合酶Ⅰ的菌株。虽然该菌株对那些可能破坏DNA的试剂异常敏感,但确实存在。
1969年冷泉港实验室的研究员凯恩斯和露西亚(Paula De Lucia)在编码polⅠ的polA基因中分离出一种具有缺陷的突变体。这个突变体(polAⅠ)缺少polⅠ活性,但它还是有活力的,从而有力地提出polⅠ实在不是DNA复制酶,而好像是在DNA损害的修复中起着主要的作用。它填充在排除错误碱基后留下的空隙。这年12月他们在Nature第224卷上发表题为《受突变影响的大肠杆菌菌株DNA聚合酶的分离》的论文。在文章一开始就写道:“科恩伯格关于在试管中能够精确复制DNA的酶的发现在分子生物学历史上是关键的一步。因为它牢固地确立了这个事实,对于能够复制全体的机制的编码只需要细胞DNA的一小部分。在那时正确地判断在体内负责DNA复制的酶是不是这个酶是不太重要的。然而,从那时以来已经累积的间接的证据表明,无论如何在细菌中这个特殊的酶是用于DNA的修复而不是它的复制。在高温下不可能复制它们的DNA的大肠杆菌和枯草杆菌的各种各样突变体,已经完全表明在不容许的温度下包含了正常的聚合酶和脱氧核苷三磷酸库,至少其中之一已经表明在高温下进行了修复合成。”[1]
在凯恩斯文章发表的同时,爱丁堡大学分子生物系教授J.Gross和 M.Gross[2]在1969年Nature第224卷上发表题为《具有影响DNA聚合酶突变的大肠杆菌菌株的基因分析》的论文。他们在文章一开始就写道:“凯恩斯和露西亚已经报告了分离的大肠杆菌突变株不同于亲代株的3大特征:在提取物中极大地减少了DNA聚合酶活性,增加了对紫外光辐射的敏感性和增加了对磺化甲基甲烷的敏感性。由凯恩斯和露西亚提出的证据表明,这些性质大致是单个损伤定位在DNA聚合酶的结构基因上的结果。我们建议相应的基因座称为polA,而在此研究的特殊突变是polAⅠ。”通过实验他们证实polA定位在染色体的metB区域,polAⅠ是琥珀型突变(amber mutant)[2]。
1969年约翰·凯恩斯从大肠杆菌中分离到的一种缺少DNA聚合酶仍然以正常的速率生长和繁殖的突变株,成为了责难DNA聚合酶和科恩伯格的有力证据。polⅠ不是主要复制酶的发现,激励起了对真实DNA复制的重新研究。在1971年,哥伦比亚大学汤姆·科恩伯格(Thomas Kornberg)和马尔克姆·杰夫塔(Malcolm Gefter)发现了两个新的聚合酶活性:DNA聚合酶Ⅱ和Ⅲ(polⅡ和polⅢ)。我们将看到polⅢ是有效的复制酶。
1970年在《自然·新生物学》(Nature:NewBiology)杂志上发表了一系列针对DNA聚合酶和科恩伯格的责难性评论。在这个时候,阿瑟· 科恩伯格的儿子汤姆·科恩伯格加入到冲突中。1970年5月汤姆因左手食指上的肿瘤恶化而使他在朱利亚音乐学院的大提琴课程不可能继续下去。其次,他就读的哥伦比亚大学,在生物学课程中,DNA聚合酶受人蔑视,他为此感到伤心。既然他无法再演奏大提琴,汤姆想知道他是否能参与在凯恩斯突变株中寻找失踪的聚合酶[3]。
结果没让人失望。汤姆在哥伦比亚大学生物系的马尔科姆·杰夫塔实验室获得了一席之地,接着在3个星期内,他就在大肠杆菌细胞中发现了一种DNA多聚酶,这种酶与他父亲已经发现的不一样。1970年9月,仅仅在他进入实验室3个月后,他向每3年一次在瑞典举办的生物化学国际会议提交了他轰动性的发现。在第2年,汤姆以研究生的身份提纯了这种新的多聚酶,并命名之DNA聚合酶Ⅱ(polⅡ)。他可以清楚地将它和DNA聚合酶Ⅰ(polⅠ)分开。在色谱分析过程中,他注意到还有一条聚合酶带从DNA聚合酶Ⅱ中完全分离出来,它所在的位置在普通细胞制剂中被DNA聚合酶Ⅰ占据,从而使之隐藏。尽管富有经验的酶学专家提出强烈的反对意见,认为这个新的染色体带像是技术假象,汤姆坚持己见,并证实它是一个不同的实体,将其命名为 DNA 聚合酶Ⅲ(polⅢ)[3]。
1971年12月汤姆·科恩伯格等[4]在《美国国家科学院院报》第68卷上发表题为《在大肠杆菌突变体中对DNA聚合酶在DNA合成中的热敏性的分析》的论文。这篇文章介绍了他们构建了一系列含有DNA合成热敏性突变(dnaA,B,C,D,E,F,G)和凯恩斯与露西亚的polAⅠ的双突变体。在每个突变体中他们测量了DNA聚合酶Ⅱ和聚合酶Ⅲ的活性。在测试的所有株中DNA聚合酶Ⅱ的活性是正常的。在dnaE基因座上在具有热敏性突变的这些株中DNA聚合酶Ⅲ的活性是特别热敏的。从这些结果他们得出结论:DNA聚合酶Ⅱ和聚合酶Ⅲ是独立的酶,DNA聚合酶Ⅲ是大肠杆菌中DNA复制最主要的酶[4]。
文章一开始他们写道:“由凯恩斯和露西亚对缺少DNA聚合酶Ⅰ活性的大肠杆菌突变体polAⅠ的分离,已经促使许多人研究这类菌株的DNA合成能力。我们和其他人已经报告了DNA聚合酶Ⅱ的提纯和特征。此外,我们又报告了在大肠杆菌中第3种DNA聚合酶(DNA聚合酶Ⅲ)的存在,但是还没有测定这些酶的生理学功能。”
他们接着指出:“没有适量DNA聚合酶Ⅰ活性的具有生存能力的细胞表明,DNA聚合酶Ⅰ不是大肠杆菌DNA复制机制的必须的组分。为了确定聚合酶Ⅱ和聚合酶Ⅲ是不是复制的主要组分,我们检验了对DNA复制温度敏感的大肠杆菌突变体的DNA聚合酶,试图使试管中的基因突变与改变的DNA聚合酶活性建立联系。我们提出的证据表明DNA聚合酶Ⅲ是在dnaE基因座上基因表达的主要产物。”[4]
文章的最后报告了他和杰夫塔在磷酸纤维素层析的基础上区别DNA聚合酶Ⅱ和聚合酶Ⅲ(图1)。其中图1(a)是从polAⅠ株的无细胞提取物中分离DNA聚合酶,图1(b)是来自野生型大肠杆菌细胞中分离DNA聚合酶。测定每个组分的DNA聚合酶Ⅰ活性,分别在35~45和12~19组分中洗脱聚合酶Ⅱ和聚合酶Ⅲ活性。典型分离的结果如图1所示,在图1(a)中polⅡ与polⅢ和polⅠ分离得很好,但在野生型细胞中polⅢ被polⅠ掩盖,见图1(b)[5]。

图1 DNA聚合酶的磷酸纤维素层析分离(纵坐标为DNA聚合酶活性,横坐标为组分数)(a)在pol AⅠ中,(b)在野生型细胞中
2 DNA聚合酶Ⅲ*的发现和全酶形式的分离
1973年6月,科恩伯格和他的同事发现DNA聚合酶Ⅲ的新形式polⅢ*和共聚物酶CopolⅢ*,提出全酶是由两个polⅢ亚基和两个CopolⅢ*亚单位组成的四聚体。1974年科恩伯格和他的同事进一步研究了DNA聚合酶Ⅲ全酶形式(holoenzyme form)的分离和性质。
2.1 DNA聚合酶Ⅲ*和共聚物酶Ⅲ*的发现
1973年6月,科恩伯格和他在斯坦福大学医学院生物化学系的同事[6]在《美国国家科学院院报》第70卷上发表题为《DNA聚合酶Ⅲ的新形式和共聚物酶复制长的单链引物模板》的论文。在摘要中介绍了DNA聚合酶Ⅲ的新形式和共聚物酶的发现。他们写道:“一种称为polⅢ星(polⅢ*)的DNA聚合酶Ⅲ的新形式已经从大肠杆菌提纯到均一性。像对polⅢ描述的一样,当从热敏的dnaE突变株分离时polⅢ*是温度敏感的。polⅢ*和polⅢ是通过凝胶过滤分离的。polⅢ*利用包含短缺口的双链体模板,具有像polⅢ一样相同的催化性质。然而,如果提供以下物品:亚精胺,引物片段和称为共聚物酶Ⅲ*(CopolⅢ*)的新蛋白质,polⅢ*可以复制长的单链模板,例如同聚物链,M13和ΦX174的病毒圆环。已提纯到均一性的CopolⅢ*没有已知的独立酶的活性,而维持合成是由polⅢ*而不是由polⅠ,polⅡ或polⅢ。”[6]
文章一开始概括地叙述了polⅢ*和CopolⅢ*发现的背景。他们写道:“在大肠杆菌提取物中发现M13和ΦX174单链圆环 DNA(single-stranded DNA,ssDNA)转变成双链复制形式(replicating form,RF)依赖于特殊的热酶系统。M13复制需要RNA聚合酶起始合成,而ΦX需要一个新型的RNA合成系统,而且也包含dnaA,dnaB,dnaC—D和dnaG基因产物。发现 M13和ΦX两者的复制需要dnaE基因产物,杰夫塔等人认为是 DNA 聚合酶Ⅲ(polⅢ)。”[6]
接着又写道:“在提纯复制 M13和ΦXDNA的酶时,我们发现提纯的polⅢ没有活性,而是这种酶的一种新型的,大概更复杂的形式,在此称之为polⅢ*的负责链的生长。一种附加的蛋白质,共聚物酶Ⅲ*(CopolⅢ*)对于polⅢ*作用是必不可少的。”[6]
接着叙述了polⅢ*和CopolⅢ*复制系统的提纯过程,通过在凝胶电泳中呈现单带,可以判断polⅢ*和CopolⅢ*是均一的,相对分子质量分别为90和77 kDa。
关于CopolⅢ*对于polⅢ*的特殊性,作者写道:“对于在一条长的单链模板,例如RNA引导的ΦX单链上polⅢ*的作用,CopolⅢ*是必不可少的(表1),但是对于具有短缺口的双链DNA的复制不是必须的。像从科恩伯格和杰夫塔的研究中所预期的一样,用单链模板polⅢ是没有活性的,而CopolⅢ*没有影响(表1),CopolⅢ*既不影响polⅠ的作用,也不影响polⅡ的作用。”[6]
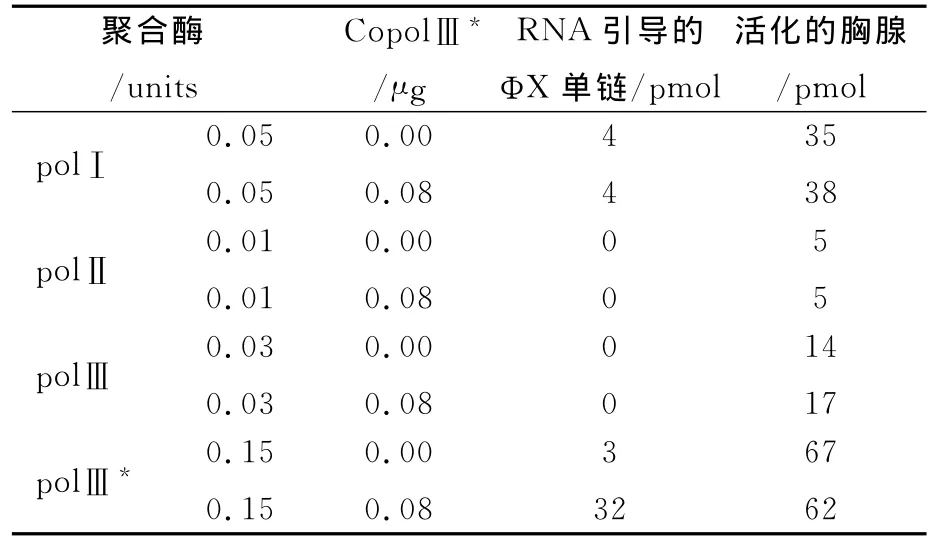
表1 模板和CopolⅢ*的酶的特殊性
通过凝胶过滤和磷酸纤维素层析,他们从polⅢ分离出polⅢ*,又从polⅢ*与polⅢ的比较中,发现复制带有缺口的双链模板显示了polⅢ*不同于polⅢ的特征。最后在讨论polⅢ*与polⅢ的联系时,他们写道:“我们对酶促M13和ΦX单链病毒圆环转变到双链复制形式的研究已经把DNA聚合酶Ⅲ的新形式显示为这个酶是造成这个复制的主要原因。这个称为polⅢ*的新酶显然是以下面方式与polⅢ相联系的:(a)dnaE基因的产物,(b)在复制具有短缺口的双链体模板具有相同的特征(用盐抑制,用于脱氧核苷三磷酸的高米氏常数Km的促进),(c)通过加热polⅢ*转变成polⅢ。polⅢ*与polⅢ的不同之处是:(a)当另一个蛋白质CopolⅢ*也存在时,polⅢ*具有复制长的单链模板的能力,(b)在琼脂糖凝胶上和在磷酸纤维素层析上分离这两种酶的物理特征。”
接着写道:“polⅢ*和CopolⅢ*也已经分离成接近均一的状态,人们有可能确定一方面是polⅢ*和polⅢ之间物理特性的不同,另一方面是polⅢ*和CopolⅢ*的相互作用。polⅢ*可能是由包括polⅢ的核心单位、具有同样尺寸的亚单位组成的全酶,或者它可能简单地是polⅢ的多聚的不对称的形式。”[6]
2.2 DNA聚合酶Ⅲ全酶形式的分离
1974年10月,科恩伯格和他在斯坦福大学医学院生物化学系的同事 W.Wickner[7]在《生物化学杂志》第249卷上发表题为《DNA聚合酶Ⅲ全酶形式的分离和性质》的论文,提出了DNA聚合酶Ⅲ的全酶形式和全酶的亚基结构。
文章一开始作者就叙述了在ssDNA复制成双链复制形式的时候,DNA聚合酶Ⅲ*(polⅢ*)和共聚物酶Ⅲ*(CopolⅢ*)是必不可少的。他们写道:“DNA聚合酶Ⅲ(polⅢ)是dnaE基因的产物,在ΦX174和M13单链病毒DNA(ssDNA)转变成双链复制形式时是没有活性的。然而,这个酶的一种比较复杂的形式DNA聚合酶Ⅲ*(polⅢ*),在附加的蛋白质共聚物酶Ⅲ*(CopolⅢ*)存在时,能够催化这个转变。用RNA引导的ssDNA模板作为开始,在这个过程中的第一步是由引物模板,亚精胺(或DNA解链蛋白),ATP和两个蛋白质polⅢ*和CopolⅢ*组成的初始复合物的形成。在这一步,ATP分裂成ADP和无机磷酸。一旦形成这个复合物,ssDNA复制成双链复制形式,就既不需要ATP也不需要CopolⅢ*。在这个反应中,polⅢ不能代替polⅢ*,在物理性质和催化性质上polⅢ*不同于polⅢ,polⅢ*好像是polⅢ的多聚形式。”[7]
在此基础上他们概括出DNA聚合酶Ⅲ(polⅢ)的3种形式,写道:“称为全酶的DNA聚合酶Ⅲ(polⅢ)的新形式已经从慢慢溶解的大肠杆菌提纯到明显的均一性。在它们的不同的物理特征和它们利用主要的单链模板的能力的基础上现在已经辨别出polⅢ的3种形式:(a)polⅢ是90 kDa亚基的二聚体,在单链环形的DNA上没有活性;(b)polⅢ*是polⅢ的较高的多聚体,只有在具有77 kDa的多肽,共聚物酶Ⅲ*(CopolⅢ*)存在时才在ssDNA上具有活性;(c)全酶是具有330 kDa的四体,由两个polⅢ亚基和两个CopolⅢ*亚单位组成,根据在附加的CopolⅢ*不存在时在ssDNA上它的活性,把全酶和polⅢ或polⅢ*区别开。通过在磷酸纤维素上层析,全酶分离成polⅢ*和CopolⅢ*;通过加热、稀释或冷却,polⅢ*转变成polⅢ。像polⅢ*一样,全酶需要ATP以便形成具有引物模板的初始复合物。”[7]
接着,他们又补充说明,现在已经发现polⅢ的第三种形式polⅢ全酶是更天然的形式。用polⅢ全酶复制ssDNA模板不需要附加的CopolⅢ*,而类似于在全酶对CopolⅢ*抗体敏感中和对ATP的依赖中由polⅢ*和CopolⅢ*的混合物共同催化。通过在磷酸纤维素上层析,全酶分离成polⅢ*,dnaE多肽和CopolⅢ*的四聚体;当分离dnaE突变株细胞时,polⅢ*是热敏的。通过冷却,稀释或温和地热处理polⅢ*能够可逆地转变成polⅢ。
亚基(subunit)是蛋白质的最小共价单位,它可以由一条多肽链或以共价键连接在一起的几条多肽链组成。作者在全酶的亚基结构一节中写道:“在1%十二烷基硫酸钠,1%β-硫基乙醇中加热全酶(10μg)并经受聚丙烯酰胺凝胶电泳(图2)。在90 kDa和77 kDa处看到两条显著的蛋白质带。给同样的凝胶加上2,4和6μg提纯的全酶。在用Coomassie蓝着色后,在580 nm处扫描凝胶,切下90和77 kDa的峰显迹并称重。当对两个峰的重量比作相对分子质量方面的校正时,观察到了两个多肽相等的克分子数的比率(表2)。假定这个90 kDa多肽是polⅢ是dnaE多肽,而77 kDa多肽是CopolⅢ*。”[7]
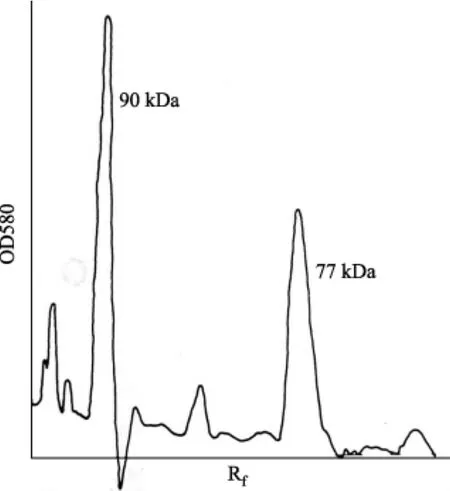
图2 全酶的凝胶电泳(Rf为相对迁移率,OD580为580 nm时的光密度)
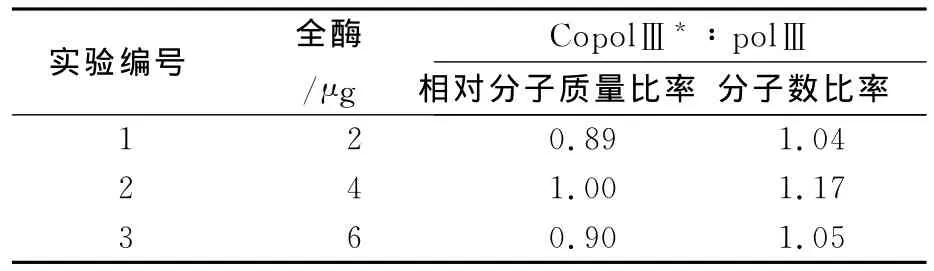
表2 在全酶中亚基的比率
为了确定全酶的相对分子质量,通过生物凝胶A-5 m柱过滤它并在甘油梯度中沉降,凝胶过滤和甘油梯度沉降两者表明,全酶相对分子质量大于过氧化氢酶(247 kDa),但是小于RNA聚合酶(490 kDa)。在这些数据和90与77 kDa多肽的等克分子数比率(表2)的基础上,假定全酶是大于二体(167 kDa)而小于六体(501 kDa),因此最可能是具有334 kDa相对分子质量由两个polⅢ多肽和两个CopolⅢ*多肽组成的四体。
关于polⅢ*和Copol*Ⅲ的结构,作者叙述道:“已经显示polⅢ*是由90 kDa的亚基组成和它在具有β-半乳糖甘酶的凝胶过滤上的混合色谱分析。这个polⅢ*凝胶过滤图像的未预料到的宽度可能反映从2个到6个或更多个亚基范围的寡聚体的存在。甘油梯度沉降对于polⅢ*给出了一个7.25 S的沉降系数。这些数据对于球形蛋白质给出了完全不同的相对分子质量;它们是与相对分子质量为360 kDa的不对称分子一致的,表明polⅢ*是由4个90 kDa多肽组成的四体。CopolⅢ*大约有77 kDa的相对分子质量,是由一个77 kDa的多肽组成的。”[7]
关于polⅢ*转变到polⅢ,作者写道:“根据复制长的单链区域模板能力局部的损失和根据它的凝胶过滤图像移动到polⅢ判断,通过温和地热处理polⅢ*能够转变到polⅢ。来自凝胶过滤和甘油梯度沉降的数据表明,polⅢ的相对分子质量是与90 kDa dnaE多肽的二聚体一致的。”
最后,作者讨论了全酶的组成,提出了全酶的假想形式。他们写道:“通过研究单链 M13和ΦX174病毒DNA的复制,我们已经观察到DNA聚合酶Ⅲ的两种新形式,它们在物理上和功能上与以前描述的polⅢ是有区别的。这个新形式具有polⅢ利用带有间隙的双链模板的能力,但是又能够复制一个延伸的单链DNA。在这个报告中叙述的形式,大概是接近于在体内起作用的聚合酶形式。它表示在细胞的平缓的溶胞产物中辨认的所有polⅢ,它是由两个90 kDa聚合酶组成的多肽和两个77 kDa共聚物酶的多肽组成的四聚体蛋白质(图3)。通过在磷酸纤维素上的层析(phosphocellulose chromatography)能够把全酶分解成这些组分。这个“核心”聚合酶活性以前被分解成称为聚合酶Ⅲ*的polⅢ多肽一个寡聚体(大概是四聚体);这个称为共聚物酶Ⅲ*的共聚物酶对于在单链上polⅢ*的活性是必不可少的。对于polⅢ*和共聚物酶Ⅲ*的复合物,我们使用全酶的术语是基于它与核心聚合酶和组成RNA聚合酶的σ亚基的复合物类似。像σ亚基一样,共聚物酶Ⅲ*适用于形成具有模板的复合物的初始阶段,但是在复制自己的过程中好像是不必要的。”[7]
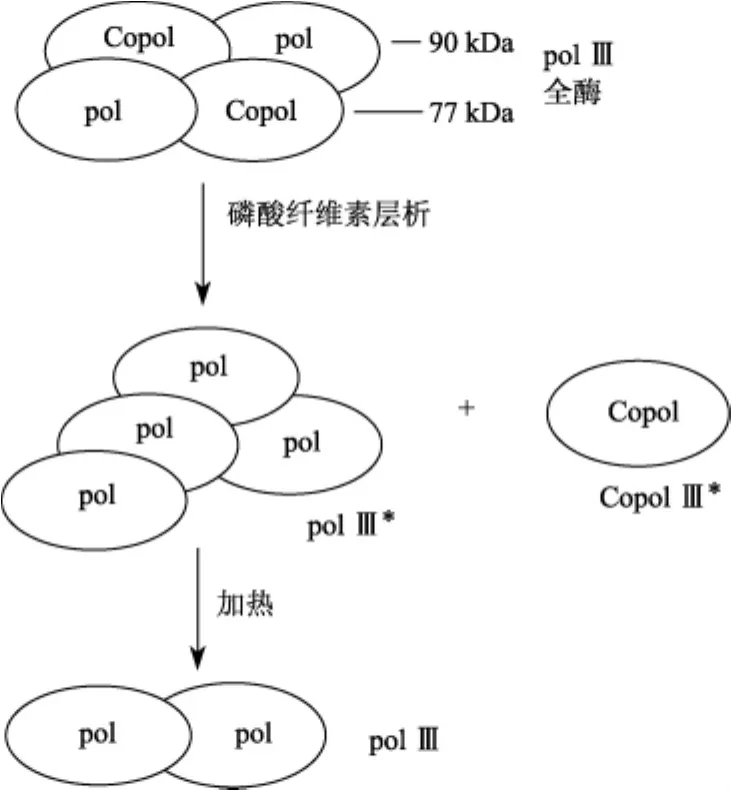
图3 聚合酶Ⅲ的假设形式
最后,作者提出了全酶的催化机制问题。他们写道:“在大肠杆菌染色体的复制中,polⅢ的几种形式的催化活性还没有弄清楚。我们对在催化中这些酶的结构的研究中发现,聚合酶和共聚物酶单位开始时在引物模板上形成一个复合物。仍然不确定的是蛋白质的化学计量法和在这个复合物中它们的空间排列,它们或者起着全酶的作用,或者以polⅢ*-CopolⅢ*对某种其他的形式起作用。即使在和缓的溶胞产物中所有聚合酶Ⅲ好像是polⅢ全酶,这个复合物仍然可能与细胞中附加的复制成分有联系。”[7]
3 DNA聚合酶Ⅲ全酶亚基的分离
1977年科恩伯格等人从大肠杆菌提纯出DNA聚合酶Ⅲ全酶,并分离出α,β,γ和δ亚基。全酶中包含的γ和δ亚基是以前没有观察到的。1979年德克萨斯医科大学生物化学和分子生物系C.Mchenry和W.Crow从提纯的DNA聚合酶Ⅲ全酶分离出ε和θ亚基。于是,DNA聚合酶Ⅲ全酶至少包含了6个不同的亚基。1982年C.Mchenry从提纯的DNA聚合酶Ⅲ全酶分离出τ亚基。1988年科恩伯格和他的同事确立了DNA聚合酶Ⅲ全酶是由10个亚基组成,除前面谈到的7个亚基外,其他3个亚基是δ′,χ和Ψ。
3.1 DNA聚合酶全酶α,β,γ和δ亚基的分离
1977年科恩伯格和德克萨斯医科大学生物化学和分子生物系C.Mchenry[8]在《生物化学杂志》第252卷上发表题为《大肠杆菌DNA聚合酶Ⅲ全酶的提纯和分离成亚基》的论文。报告了他们从大肠杆菌HMS-83中已经提纯出DNA聚合酶Ⅲ全酶,用于试验时,把大肠杆菌噬菌体G4单链DNA转变成双链复制形式。通过磷酸纤维素层析从全酶分离出相对分子质量分别为140,40,52和32 kDa的α,β,γ和δ亚基,并指出这个α亚基是DNA聚合酶Ⅲ,dnaE基因的产物,而亚基β类似于共聚物酶Ⅲ*(CopolⅢ*)。这个全酶与以前从大肠杆菌H560中分离的,在磷酸纤维素层析上能够分离成包含polⅢ亚基和CopolⅢ*的共聚物类似。但是,全酶中包含的γ和δ亚基是以前没有观察到的。[8]
在“全酶的组分”一节中叙述了全酶的分离和全酶组分亚基的识别。他们使用聚丙烯酰胺凝胶电泳分离聚合酶并测定其相对分子质量。他们在“全酶的SDS-聚丙烯酰胺凝胶电泳”一段中写道:“用重氮化的[35S]磺酸标记全酶(0.2μg),变性后,在聚丙烯酰胺凝胶上电泳显示出4条蛋白质带。已经证明它们是为引导G4模板延伸所需要的组分。这个140 kDa蛋白质(α)大概是polⅢ,dnaE基因的产物。其他人已经提纯polⅢ,发现它基本上是包含了这个量值的组分。第2个40 kDa的亚基β可能是以前分离的CopolⅢ*,被认为是77 kDa的多肽。两个组分γ和δ(相对分子质量分别为52和32 kDa)已经提纯为一个α和β作用所需要的复合物。基于对α-,β-,γ-和δ-带的光密度计扫描,这个全酶制剂至少60%的提纯是这4个亚基的含量。还存在83和25 kDa两个附加的蛋白质,在提纯到这个程度的整个过程中它们仍然与全酶牢固地联系在一起,在甘油梯度中带着全酶活性沉降。”[8]
最后,他们指出:“DNA聚合酶Ⅲ全酶不是一个物理上的整体,它的活性来源于4个分离的蛋白质的混合物。”[6]
3.2 DNA聚合酶全酶ε,θ,τ亚基的分离
1979年德克萨斯医科大学生物化学和分子生物系C.Mchenry等[9]在《生物化学杂志》第254卷上发表题为《大肠杆菌DNA聚合酶Ⅲ亚基的提纯和识别》的论文。文章指出,这个DNA聚合酶Ⅲ全酶的核心已经从大肠杆菌HMS-83中提纯28 000倍到97%的均一性。这个酶包含相对分子质量分别为140,25和10 kDa的α,ε和θ亚基。这个α亚基以前已经表示为DNA聚合酶Ⅲ和更为复杂的DNA聚合酶Ⅲ全酶两者的组分。在此证实ε和θ亚基也是DNA聚合酶Ⅲ全酶的亚基。于是,DNA聚合酶Ⅲ全酶至少包含了6个不同的亚基。在这篇论文中作者报告了DNA聚合酶Ⅲ提纯到均一性和DNA聚合酶Ⅲ和DNA聚合酶Ⅲ全酶两者的两个补充亚基的识别。[9]
1982年C.Mchenry[10]在《生物化学杂志》上发表题为《DNA聚合酶Ⅲ′的提纯和特征与DNA聚合酶Ⅲ全酶的τ亚基的识别》的论文。在这篇论文中作者指出:“DNA聚合酶Ⅲ′是DNA聚合酶Ⅲ的新形式,已经从大肠杆菌K12株提纯15 000倍到90%的均一性。DNA聚合酶Ⅲ′是DNA聚合酶Ⅲ全酶4个亚基的组件,它具有在核心DNA聚合酶Ⅲ和全酶之间的功能和物理性质。在变性条件下完成的聚丙烯酰胺凝胶电泳表明DNA聚合酶Ⅲ′是DNA聚合酶Ⅲ的α,ε和θ亚基和新测定的DNA聚合酶全酶的τ亚基(Mτ=83 kDa)的复合物。通过凝胶过滤和磷酸纤维素层析从DNA聚合酶Ⅲ′分离DNA聚合酶Ⅲ。所有酶形式能够利用包含短间隙的双链体模板。DNA聚合酶Ⅲ′像DNA聚合酶Ⅲ全酶一样,在存在5 mM(毫摩尔)亚精胺时能够在长的单链模板上合成DNA;而DNA聚合酶Ⅲ不能够。在G4自然复制系统中DNA聚合酶Ⅲ′没有作用,而DNA聚合酶Ⅲ全酶是有活性的。相对分子质量和亚基的化学计量法确定DNA聚合酶Ⅲ′包含两个单位的核心DNA聚合酶Ⅲ和两个τ亚基。”[10]
作者又指出:“在这篇论文中,我报告了一种同样定义的,其功能和结构介于DNA聚合酶Ⅲ和DNA聚合酶Ⅲ*之间的聚合酶形式的提纯和特征。它的提纯已经导致新的全酶亚基τ的测定和这个亚基对全酶反应的贡献部分的理解。”[10]
在介绍了DNA聚合酶Ⅲ′的提纯后,作者叙述了新的全酶亚基τ的测定。他写道:“变性DNA聚合酶Ⅲ′的级分Ⅵ(10μg),并在SDS 聚丙烯酰胺凝胶上电泳,出现为140,83,25和10 kDa的4条主要的蛋白质带(图4)。最大的和两个最小的组分是核心DNA聚合酶Ⅲ的亚基,称为τ的第4个组分(83 kDa)不是核心聚合酶的一部分。光密度计扫描表明α∶τ的比率是1∶1.1,在不同的制剂中两个较小的亚基ε和θ的比率在1和2之间改变。由于结合到很不同的相对分子质量的蛋白质的染料的变化,我将不测定ε和θ的化学计量。光密度计扫描也表明DNA聚合酶Ⅲ制剂提纯到90%”。[10]

图4 SDS 聚丙烯酰胺凝胶电泳的光密度扫描(纵坐标为光密度,横坐标为组分数)
接着作者叙述了在DNA聚合酶Ⅲ全酶中τ亚基的存在,他写道:“DNA聚合酶Ⅲ′的τ亚基存在于高度提纯的DNA聚合酶Ⅲ全酶中(图4)。τ与存在于所有全酶制剂中的83 kDa组分一起迁移。使用的全酶级分是可能得到的高度提纯的制剂。它用的提纯程序显然不同于DNA聚合酶Ⅲ′使用的提纯程序。基于这个信息和下面要指出的功能信息,τ被确定为DNA聚合酶Ⅲ全酶的亚基。于是,DNA聚合酶Ⅲ′是4个全酶亚基的组件。”[10]
3.3 DNA聚合酶Ⅲ全酶的组成
1988年,科恩伯格和他在斯坦福大学医学院生物化学系的同事H.Maki等[11]在《生物化学杂志》第249卷上发表题为《大肠杆菌DNA聚合酶Ⅲ全酶Ⅳ:全酶是一个具有成对活性部位的不对称的二聚体》的论文。一开始他们概述了DNA聚合酶Ⅲ全酶的组成。他们指出:polⅢ*是只缺少辅助的β亚基的大肠杆菌DNA聚合酶Ⅲ全酶的组件,已经通过改进的程序提纯到均一。这个组件是由9个不同的多肽组成的。在凝胶过滤上这个组件相当于大约为800 kDa的粒子,表示每种亚基含有两个。保留β亚基(37 kDa)以及在polⅢ*中的所有亚基;全酶的相对分子质量估计是900 kDa。基于这些数据,断定polⅢ全酶是具有成对的polⅢ核心活性部位和两个不同组的辅助亚基的一个不对称的二聚体,以便用来达到前导链和后续链二者基本上并行复制。
接着,作者介绍了DNA聚合酶Ⅲ的三个分离形式,写道:“DNA聚合酶Ⅲ的三个分离形式很可能是全酶的组合件。由α,ε和θ亚基组成的核心(polⅢ)有一个165 kDa估计的相对分子质量。它缺少连续合成的能力,但是保持了催化功能:在α亚基中聚合酶活性和在ε亚基中3′→5′外切核酸酶活性。第二种形式polⅢ′,包括核心和τ亚基。由于410 kDa的反常的量值,对于这个复合物提出二聚体结构。polⅢ*是缺少β的全酶,这个亚基对于连续性是关键的。基于360 kDa或540 kDa的相对分子质量,polⅢ*好像是polⅢ′和其他辅助亚基的混合物。解释全酶为何具有与polⅢ′一样相同的沉降系数是困难的。由于β亚基的低亲和性,甚至在ATP稳定的存在中全酶与polⅢ*的联系也是不清楚的。”[11]
在这篇文章中,他们报告了polⅢ*和全酶的新的提纯程序,并弄清楚两种复合物的一些特征。polⅢ*大约具有800 kDa的相对分子质量,包含9个不同的很可能是相同相对分子质量的多肽。全酶分离为具有两个β二聚体的polⅢ*的复合物,具有900 kDa的相对分子质量。基于这些量值和从亚基重新构成的复合物的分解,断定polⅢ*和全酶两者是具有辅助亚基非对称分布的二聚型(图5)。这种结构可能提供在复制叉处具有催化前导链和后续链并行合成的能力。
4 DNA聚合酶Ⅲ全酶的结构与功能
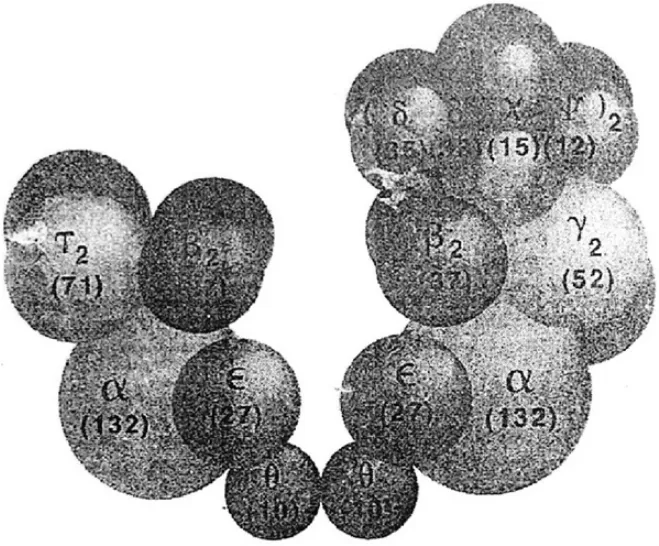
图5 polⅢ全酶假定的结构
1995年康奈尔大学微生物系的Z.Kelman教授等[12]在《生物化学年度评论》中发表题为《DNA聚合酶Ⅲ全酶:染色体复制机制的结构和功能》的论文,全面评述了DNA聚合酶Ⅲ全酶的结构与功能。DNA聚合酶Ⅲ全酶是能够进行引物链的延伸并完成DNA的前导链和后续链合成的酶。全酶是一个多亚基酶。他们发现全酶在DNA合成上特别迅速,大约每秒合成750个核苷酸,与在大肠杆菌中观察到的复制叉运动速率是一致的,比polⅠ的每秒合成10~20个核苷酸的速率快。这样快的速率来自于全酶的高度持续的合成能力。
全酶合成DNA的高速度和连续性是与它由多亚基组成分不开的。在“全酶粒子”一节中作者指出:“现在已经完成编码全酶10个亚基的所有基因的识别,这些蛋白质的过度表达和提纯,以及从它们重新构成全酶。在表3中按顺序列出了10个不同的亚基,表明亚基以各种各样全酶的组件形式存在。核心聚合酶由α,ε和θ亚基组成。polⅢ′组件由两个核心酶和一个τ二聚体组成。在一个分子结构中两个聚合酶的存在支持这个假设,为了协调一个双链体染色体的双链的复制,复制的聚合酶要成对地起作用。polⅢ*组件包含了9个不同的亚基,它只缺少β亚基。基于把亚精胺,ssDNA结合蛋白(SSB蛋白),乙醇加到试验中或者把盐加到实验中能够识别每个组件的聚合酶活性。一般来说,当聚合酶亚基的复杂性增加时,聚合酶变得更有连续性,但是全酶很高的速度和连续性绝对需要β夹子(clamp)。这5个亚基的γ复合物是把水解的ATP结合到引物DNA上的载荷β夹子的媒介物。”[12]
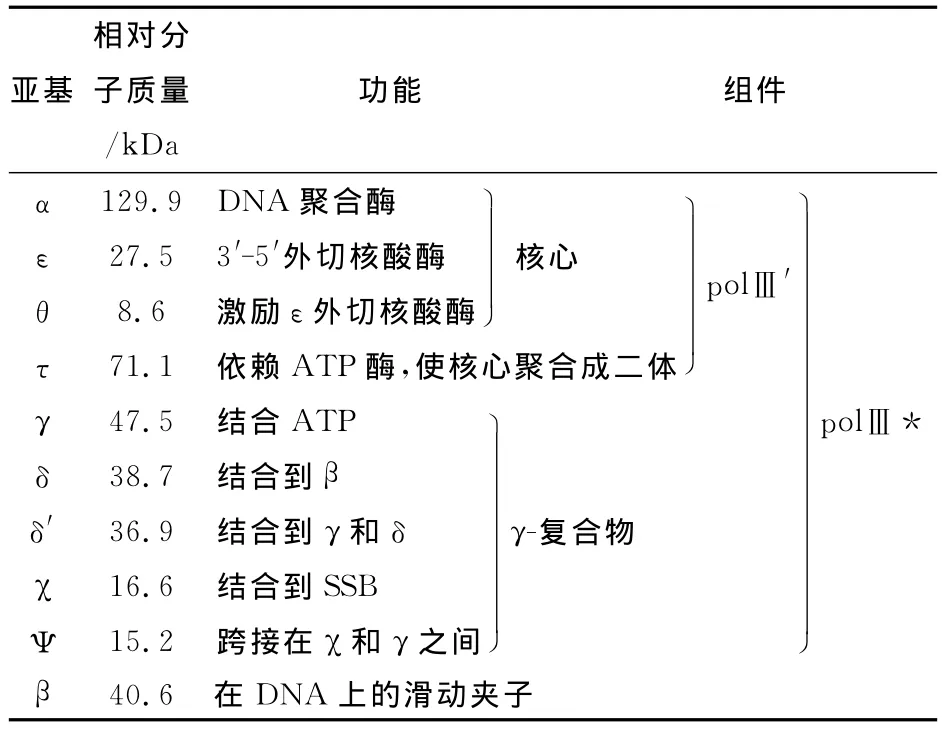
表3 DNA聚合酶Ⅲ全酶的亚基和组件
4.1 核心聚合酶
在1995年Kelman文章的“核心聚合酶”一节中介绍了核心聚合酶的构成与功能。他写道:“核心包含DNA聚合酶和校对外切核酸酶活性。在这个细胞中大约有40个核心分子,因此只有一半组装进全酶。核心的3个亚基牢固地联结在一起,缺少变性不能分解。通过基因的使用提供了各个亚基。α的研究表明它是DNA聚合酶,具有每秒合成8个核苷酸的速率,但是它缺少外切核酸酶活性。这个分离的ε亚基是强有力的3′→5′外切核酸酶。这个α和ε亚基形成牢固的1∶1的复合物,导致聚合酶活性和外切核酸酶活性两者的增加。由ε水解ssDNA的速率类似于由核心水解ssDNA的速率,但是对于有效的活性由ε水解双链DNA需要α。大概识别α位置的引物模板携带着与配对碱基3′端接触的ε。除了在错配T-G碱基对上ε活性的微小激励外,还发现了θ的功能。这个θ亚基结合ε而不是α,从而提出在核心中α-ε-θ的线型排列,而结构分析显示了每个亚基的单个拷贝。”[12]
接着作者指出了核心酶合成DNA的速率和连续性。他写道:“核心以大约每秒20个核苷酸的速率合成DNA,具有复制11个核苷酸的连续性,类似于polⅠ。然而在单个ssDNA病毒引物模板上,核心是已知的最微弱的聚合酶。无论怎样加上许多核心或等待多长时间,核心都不能延伸环绕自然模板的独特的完整的引物圆环。大概某些DNA结构阻碍由核心引起的链的延伸。”[12]
作者又补充写道:“在核心的附属蛋白质存在时,核心成为了最快的聚合酶。在ε不存在时,α受附属蛋白质的激励,但是持续合成能力降到500~1 500个核苷酸,而实际的速率是核心速率的一半。带有附属蛋白质的这个αε复合物具有像核心一样的复制速率和连续性。因此,ε对全酶的速率和持续作用能力有影响,不只是对精确度有影响。在其他方面,θ对αε的效率没有影响。”[12]
4.2 ε亚基的外切核酸酶活性
1984年加利福尼亚大学分子生物系教授R.Scheuermann和 H.Echols[13]在《美国国家科学院院报》上发表题为《DNA复制的一个独立编辑的外切核酸酶:大肠杆菌DNA聚合酶Ⅲ全酶的ε亚基》论文。文章一开始就阐述了确保复制精确度的机制。写道:“大肠杆菌基因组的复制是一个特别精确的过程。每个碱基复制错误的频率一般是10-9~10-10。这样高的精确度被认为是通过多级过程出现的:(1)在初始的5′→3′结合中互补碱基的选择;(2)在生长点非互补碱基的外切核酸溶解的3′→5′删除;(3)复制后错配的修复。这些步骤的总和能够达到观察的精确性。DNA聚合酶Ⅲ(polⅢ)全酶是在大肠杆菌中链的延伸中包含的主要的酶,因此很可能是精确度的主要决定因素。为了研究在DNA复制中的保真机制和探索控制精确度的可能性,我们一直试图确定polⅢ亚基对碱基选择和删除的贡献。”[13]
接着,他们分析了全酶核心的活性,并提出了3′→5′外切核酸酶活性定位在哪个亚基上的问题。写道:“polⅢ全酶至少有7个亚基:α,ε,θ,τ,γ,δ和β。以自然形式制备的polⅢ全酶的最小组件是包含α,ε和θ亚基的polⅢ核心;α是dnaE基因的产物,而ε是dnaQ基因的产物。polⅢ核心带有polⅢ全酶的聚合酶和3′→5′外切核酸酶活性。具有用聚丙烯酰胺凝胶电泳分离的亚基的酶测定表明这个大的α亚基具有聚合酶活性,也可能带有3′→5′外切核酸酶活性。然而,已经从观察到dnaQ基因中增变突变(mutator mutation)使polⅢ全酶在删除的外切核酸酶中存在缺陷,推断在3′→5′外切核酸酶(和复制精确度)中ε的重要作用。”[13]
随后,他们叙述了通过ε亚基的过度表达和提纯,证实ε亚基具有外切核酸酶活性。写道:“为了确定在由polⅢ外切核酸裂解删除中ε的作用,我们要从多亚基的polⅢ全酶的其他亚基分别地研究ε。为了提供过量的ε和使它的提纯容易,我们使用了过量的菌株,在强有力启动子,噬菌体λ的左侧区域转录启动子和有效的核糖体结合区域的控制下表达dnaQ基因。我们已经把polⅢ的ε亚基提纯到均一性。我们发现ε带有类似于提纯的polⅢ核心酶特征的3′→5′外切核酸酶活性。于是,polⅢ全酶的删除和聚合作用活性存在于不同的亚基中。”[13]
4.3 β亚基滑动夹子的功能
1992年5月,洛克菲勒大学分子生物物理实验室X.Kong等[14]在Cell第69卷上发表题为《大肠杆菌DNA聚合酶Ⅲ全酶的β亚基的三维结构:滑动的DNA夹子》的论文。说明当把其余的机构夹到模板上时,β亚基是一个沿着DNA滑动的环形蛋白质,其他亚基共同起着把这个环装配到DNA上的撮合者的作用。在文章的摘要中他们首先概述了这个结构特点,写道:“已经以2.5Å的分辨距离确定了DNA聚合酶Ⅲ全酶的β亚基的晶体结构。β亚基的二聚体(M=2×40.6 kDa,2×366个氨基酸残基)形成了用12个能环绕双链体DNA的α螺旋嵌入的环形结构。这个结构是高度对称的,具有包含三个相同拓扑结构域的每个单体。这些螺旋的电荷分布和定位表明通过形成能够在DNA上滑动的牢固的夹子,这个分子起着生物化学的作用。提出在β亚基和增殖细胞核抗原(PCNA,真核生物聚合酶δ和ε持续合成能力因子),和噬菌体T4DNA聚合酶的基因45蛋白质之间可能的结构联系。”[14]
在文章的“引言”中,概述了polⅢ全酶各个亚基的作用。写道:“未受损害的polⅢ全酶至少具有10个不同的蛋白质亚基(α,ε,θ,τ,γ,δ,δ′,χ,Ψ 和β)。这个α亚基完成催化的聚合酶功能,而ε亚基是3′→5′外切核酸酶。这个包含了全酶的α,ε,和θ的3个亚基核心聚合酶组件,虽然它能够插进短的单链区域,但不可能独自地连续地起作用。按照核心聚合酶与β亚基和5个蛋白质的γ复合物(γ,δ,δ′,χ和 Ψ)的混合能够重新构成全酶的高度连续性特征。这个连续性聚合酶的重组是在两个不同的阶段进行的(图6):在第一阶段,这个γ复合物水解ATP,以便把β亚基转移到引物模板;在第二阶段,具有在DNA上的β亚基的核心聚合酶组件形成连续性的聚合酶。于是,β亚基把值得注意的连续性给予核心聚合酶。对为了组装连续性的聚合酶所需要的最小亚基数的研究表明,为了把β从溶液转移到引物模板只需要γ复合物的γ和δ亚基。对于连续的聚合作用需要作为αε复合物的两个亚基α和ε。只要在DNA上的β亚基使αε聚合酶具有完整的连续性性能,γ复合物便接着被排除。”[14]
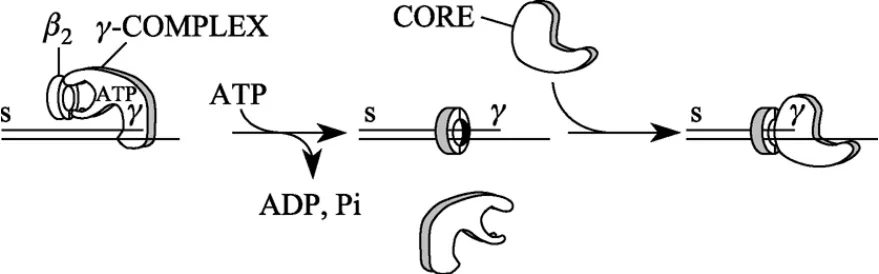
图6 连续性聚合酶的两阶段组合
下面叙述了在DNA复制过程中β亚基,γ复合物与引物模板之间的相互作用。写道:“一旦γ复合物完成把β亚基夹紧在DNA上的操作,β亚基就很牢固地被束缚在那里,它就不能很容易地与圆环DNA分离。然而,已经显示出它沿着双链体DNA自由地滑动,与它把聚合酶核心束缚到模板上,在复制中与聚合酶一道运动,起着夹子的作用是一致的。使用限制性内切核酸酶的实验已经揭示,在β亚基被夹紧之后,如果圆环DNA被切割,β亚基就滑动到断裂位置并跌落下来,完全与DNA分离。这些与在同样的研究中有关的实验所表明的,与位置特异性结合蛋白,诸如转录因子或核酸酶形成特殊的氢键或其他的与DNA稳定的相互作用不同,β亚基被束缚到DNA上主要是由于它的拓扑结构而不是稳定的相互作用。已经提出β亚基可能形成一个闭合的环,γ复合物的一种作用可能是打开然后关闭这个环绕DNA的环,有效地把β亚基夹在DNA上。”[14]
接着介绍了β亚基的三维结构。写道:“作为朝着polⅢ全酶连续性质的分子基础的详细理解的一步,我们已经使β亚基结晶,通过在2.5Å分辨距离下的X射线衍射确定了它的三维结构。在与以前的推测一致感到满意时,我们发现这个结构的确是闭合的环,整个形状类似于油煎圈饼或环形线圈的形状(图7)。”[14]
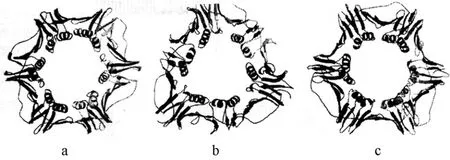
图7 滑动的β亚基的三维结构
在“结果和讨论”一节中,作者具体介绍了β亚基二聚体的结构。写道:“这个β亚基在晶体中形成头 尾二聚体,与以前观察的这个在溶液中分离的蛋白质是二聚体一致的。这个二聚体的多肽主链的图像显示在图7a中。总的结构是直径大约为80Å的星形环在中央具有一个直径大约为35Å的孔洞。这个双重的二聚体的轴垂直于环面,环的厚度大约是双链体B 形式DNA一整圈的大小(约34Å)。”[14]图7b表示T4噬菌体DNA夹是gp45蛋白的三聚体。1994年 X.Kong等[15]又在Cell第79卷上发表题为《真核生物DNA聚合酶持续合成能力因子细胞周期蛋白(PCNA)的晶体结构》的论文,报告了他们的真核滑动DNA夹子是PCNA蛋白的三聚体(图7c)。
4.4 γ复合物的结构与功能
在1995年Kelman文章的“作为复制机器的DNA聚合酶Ⅲ全酶”一节中介绍了γ复合物的构成与功能。叙述了β圆环从γ复合物到核心的转换。写道:“为了持续合成这个γ复合物必须把环形β亚基结合到引物端上的组件,而核心必须与β亚基相互作用。因为核心和γ复合物两者都识别引物模板结合处,它们可能与β环的同一个面相互作用(图8)。比较来自7种不同的细菌的基因序列编码表明,多数保守的残基只处在一个面上:这个面包含两个C端。”[10]如图8所示,全酶包含结合到τ二聚体两个核心聚合酶和一个γ复合物夹子装载器。这个γ复合物与β二聚体的C端相互作用,大概β的这个面朝着引物位置。像γ复合物一样,核心与β上相同的C端残基相互作用。因此,这个γ复合物在把β装载到DNA上之后,这个核心可能进入带有β夹子的位置。在这个全酶中通过与τ的相互作用使γ复合物抓牢带有核心和β的DNA。[12]
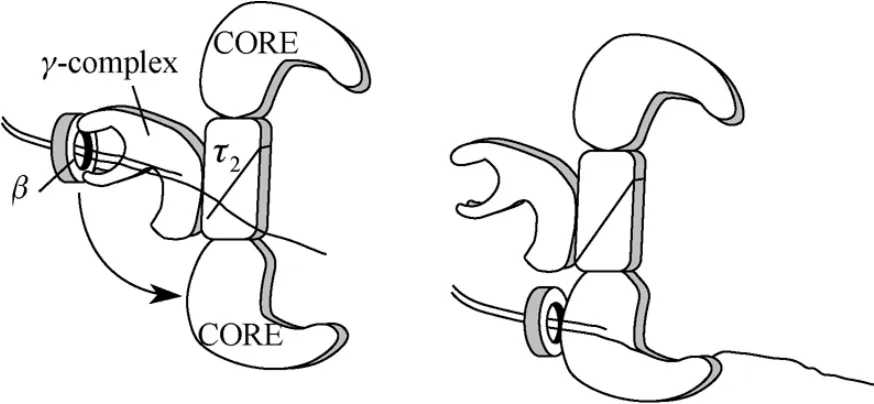
图8 核心和γ复合物与β环同一面的相互作用
作者又问道:“为何核心和γ复合物在β上具有交叠结合位置?”他回答说:“这个γ复合物不只是把β装载到DNA上,而且也从DNA上卸下β夹子。因此,这个双重性的装置能够确保当核心用β延伸DNA时,它防止γ复合物从DNA上卸下β。”[12]
作者接着写道:“使用组件(γ复合物,核心和β)的研究引起了这种想法,因为γ复合物起催化作用,在链的延伸过程中在DNA上只存在核心和β。事实上γ复合物和核心在β二聚体上交叠结合的位置是与这个观点一致的。然而,使用整个全酶的研究表明,这个γ复合物在DNA上仍然具有核心和β。在这个全酶中,τ作为核心和γ复合物之间的桥梁,使它们保持在一起。像图8所示,这种结构可以容许β从γ复合物位置改变到核心位置。把γ复合物的催化夹子装载活性放在复制叉处的适当位置,通过与全酶不变的联系,对于在后随链上许多初始事件将是有利的。”[12]
(2012年1月3日收到)
[1]DE LUCIA P,CAIRNS J.Isolation of an E.coli strain with a mutation affecting DNA polymerase[J].Nature,1969,224(5225):1164-1166.
[2]GROSS J,GROSS M.Genetic analysis of an E.coli strain with a mutation affecting DNA polymerase[J].Nature,1969,224(5225):1166-1168.
[3]阿瑟·科恩伯格著.酶的情人:一位生物化学家的奥德赛 [M].崔学军,等,译.上海:上海科学技术出版社,2004:245.
[4]GEFTER M L,HIROTA Y,KORNBERG T,et al.Analysis of DNA polymerasesⅡandⅢin mutants ofEscherichiacolithermosensitive for DNA synthesis[J].Proc Nat Acad Sci USA,1971,68(12):3150-3153.
[5]WEAVER R F.Molecular biology[M].3th ed.Boston:McGraw-Hill,2004:687-691.
[6]WICKNER W,SCHEKMAN R,GEIDER K,et al.A new form of DNA polymeraseⅢand a copolymerase replicate a long,single-stranded primer-template[J].Proc Nat Acad Sci USA,1973,70(6):1764-1767.
[7]WICKNER W,KORNBERG A.A holoenzyme form of deoxyribonucleic acid polymeraseⅢisolation and properties[J].The Journal of Biological Chemistry,1974,249:6244-6249.
[8]MCHENRY C,KORNBERG A.DNA polymerase ofEscherichia colipurification and resolution into subunits[J].The Journal of Biological Chemistry,1977,252:6478-6484.
[9]MCHENRY C S,CROW W.DNA polymeraseⅢofEscherichia colipurication and identification of subunits[J].The Journal of Biological Chemistry,1979,254:1748-1753.
[10]MCHENRY C S.Purification and characterization of DNA polymeraseⅢ.Identification ofτas a subunit of the DNA polymeraseⅢ holoenzyme[J].The Journal of Biological Chemistry,1982,257:2657-2663.
[11]MAKI H,MAKI S,KORNBERG A.DNA polymeraseⅢholoenzyme ofEscherichiacoliⅣ.The holoenzyme is an asymmetric dimer with twin active sites[J].The Journal of Biological Chemistry,1988,263:6570-6578.
[12]KELMAN Z,O′DONNELL M.DNA polymeraseⅢholoenzyme:structure and function of a chromosomal replicating machine[J].Annu Rev Biochem,1995,64:171-200.
[13]SCHEUERMANN R H,ECHOLS H.A separate editing exonuclease for DNA replication:theεsubunit ofEscherichiacoliDNA polymerase Ⅲ holoenzyme[J].Proc Nat Acad Sci USA,1984,81:7747-7751.
[14]KONG X,ONRUST R,O'DONNELL M,et al.Three-dimensional structure of theβsubunit of E.coli DNA polymeraseⅢholoenzyme:a sliding DNA clamp[J].Cell,1992,69(3):425-437.
[15]KRISHNA T,KONG X,CARY S,et al.Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA.1994,79(7):1233-1243.
Discovery of Function and Structure of Deoxyribonucleic Acid PolymeraseⅢHoloenzyme
XIANG Yi-he
Professor,DepartmentofPhysics,TsinghuaUniversity,Beijing100084,China
The process of finding the structure and the function of DNA polymeraseⅢholoenzyme are introduced.The key events include that the discovery of DNA polymeraseⅢandⅢ*,the proposition of DNA polymeraseⅢholoenzyme,the separation of holoenzyme subunit,the function of core polymerase,the 3→5 exonuclease activity ofεsubunit,the function ofβsubunit sliding clamp andγcomplex.
DNA polymeraseⅢ,DNA polymeraseⅢ*,CopolymeraseⅢ*,core polymerase,εsubunit,βsubunit sliding clamp,γ complex
10.3969/j.issn.0253-9608.2012.04.008
(编辑:段艳芳)
猜你喜欢
中等数学(2022年1期)2022-06-05
世界最新医学信息文摘(2020年68期)2020-12-25
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
烟台大学学报(自然科学与工程版)(2020年1期)2020-02-08
中学数学研究(江西)(2018年8期)2018-08-30
中学数学研究(广东)(2018年23期)2018-03-05
初中生世界·九年级(2017年9期)2017-10-13
中国病理生理杂志(2017年2期)2017-01-17
