
PPIs抑制PI3K/Akt/mTOR信号通路逆转胃癌细胞的化疗多药耐药*
2012-05-15 07:35:04李建琦许春红邹晓平
胃肠病学 2012年10期
李建琦 陈 敏 张 松 王 军 许春红 邹晓平
南京大学医学院附属鼓楼医院消化科(210008)
胃癌是消化系统最常见的恶性肿瘤之一,早期诊断率低,多数患者确诊时已为进展期胃癌,单纯手术治疗常难以根治,化疗在综合治疗中发挥重要作用,可延长患者术后生存期[1]。然而,诸多因素可导致肿瘤细胞产生多药耐药(multidrug resistance,MDR),从而影响化疗效果。ATP-结合盒(ATP-binding cassette,ABC)转运蛋白超家族成员介导的药物外排作用是导致MDR的重要原因,P糖蛋白(P-glycoprotein,P-gp)和多药耐药蛋白 1(multidrug resistance protein 1,MRP1)为该家族中与MDR关系最为密切的两个重要成员[2,3]。
缺氧和酸化是肿瘤细胞微环境最主要的特征。肿瘤细胞内外存在pH梯度,胞外pH值偏酸性,胞内pH值偏中性,与肿瘤细胞的存活、生长、侵袭以及对化疗药物耐药有关[4]。肿瘤细胞内无氧糖酵解旺盛,产生大量H+,但仍能维持细胞内外pH梯度,空泡型质子泵(vacuolar H+-ATPases,V-H+-ATPases)在其中起关键调节作用;肿瘤细胞的空泡型质子泵活性极强,在细胞膜和胞内酸性囊泡膜上高度表达[5,6]。质子泵抑制剂(PPIs)预处理能增强肿瘤细胞的化疗敏感性,与其抑制空泡型质子泵活性,逆转肿瘤细胞内外pH梯度,显著增加化疗药物在胞质内的聚集有关[7]。因此,PPIs有望作为一种新型化疗增敏剂和耐药逆转剂应用于临床。
已有研究[8]证实PPIs系通过抑制空泡型质子泵,逆转细胞内外pH梯度而增强胃癌细胞的化疗敏感性,本课题组的前期实验发现该作用与抑制MDR蛋白P-gp、MRP1表达有关。胞外酸化环境可活化胞内PI3K/Akt信号通路[9],而 PPIs可显著升高胞外pH值,推测PPIs可能对胞内PI3K/Akt信号通路产生抑制作用。哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)不仅是PI3K/Akt信号通路的下游分子[10],亦为缺氧诱导因子-1α(HIF-1α)的上游关键调节分子[11],因此推测PPIs可能通过抑制空泡型质子泵,逆转细胞内外pH梯度,进而抑制胞内PI3K/Akt/mTOR信号通路,下调HIF-1α和MDR蛋白P-gp、MRP1表达,从而提高胞内化疗药物浓度,逆转胃癌细胞对化疗药物的MDR。本研究对上述推测进行了验证。
材料与方法
一、胃癌细胞株、药物和试剂
人胃腺癌细胞株SGC7901敏感株由南京鼓楼医院肿瘤科惠赠,SGC7901多药耐药株(SGC7901/MDR)由南京鼓楼医院消化科实验室以阿霉素和顺铂诱导产生。阿霉素对SGC7901和SGC7901/MDR的 IC50分别为(0.654 ±0.103)μg/mL 和(3.203 ±0.302)μg/mL(P <0.05)。
注射用埃索美拉唑钠(阿斯利康制药有限公司),注射用泮托拉唑(德国 Nycomed GmbH);LipofectamineTM2000、DAPI、荧光二抗(invitrogenTM,Life Technologies Corporation),V-H+-ATPases siRNA(Silencer®Select Pre-Designed & Validated siRNA,ambion®,Life Technologies Corporation),雷帕霉素(LC Laboratories),V-H+-ATPases抗体 (Abnova Corporation),P-gp抗体、MRP1抗体、HIF-1α抗体、hamartin(TSC1)抗体、tuberin(TSC2)抗体、Rheb抗体(Abcam plc.),PI3K p110α 抗体、Akt抗体、mTOR抗体、phospho/TSC2(Ser939)抗体、phospho/TSC2(Thr1462)抗体、phospho-(Ser/Thr)Akt底物抗体(Cell Signaling Technology,Inc.),山羊抗鼠、山羊抗兔二抗(杭州联科生物技术有限公司)。
二、实验方法
1.细胞培养:SGC7901/MDR、SGC7901细胞以含10%胎牛血清的RPMI-1640培养基按贴壁方法培养于37℃、5%CO2孵箱内,SGC7901/MDR细胞培养基内含1000 ng/mL阿霉素以维持其MDR表型。
2.PPIs预处理:取对数生长中期 SGC7901/MDR、SGC7901细胞,在pH 6.65的未缓冲培养体系中(弱酸环境可促进PPIs活化)以不同浓度埃索美拉唑或泮托拉唑(0、10、20、50、80、100μg/mL)预处理24 h,收集细胞。
3.siRNA干扰:取对数生长中期 SGC7901/MDR细胞接种于6孔板,以含10%胎牛血清的RPMI-1640培养基按贴壁方法于37℃、5%CO2孵箱内培养24 h,更换为无血清培养基饥饿12 h,于无血清培养基中加入LipofectamineTM2000和V-H+-ATPases siRNA处理24 h,收集细胞。
4.雷帕霉素阻滞:取对数生长中期SGC7901/MDR细胞接种于6孔板,以含10%胎牛血清的RPMI-1640培养基按贴壁方法于37℃、5%CO2孵箱内培养24 h,更换为无血清培养基饥饿12 h,去除培养基,以不同浓度雷帕霉素(0、20、40、80μg/mL)处理24 h,收集细胞。
5.蛋白质印迹法:取上述经不同方式处理的细胞,加入预冷细胞裂解液置冰上裂解30 min,4℃12000 r/min离心(离心半径7.5 cm),取上清液行蛋白定量。取30μg总蛋白,SDS-PAGE电泳分离,转移至PVDF膜,5%脱脂牛奶封闭2 h,加入相应一抗[V-H+-ATPases、PI3K p110α、Akt、mTOR、HIF-1α、TSC1、TSC2、Rheb、phospho/TSC2(Ser939)、phospho/TSC2(Thr1462)、phospho-(Ser/Thr)Akt底物:1∶1000;P-gp:1∶500;MRP1:1∶50),4 ℃孵育过夜。以含Tween 20的TBST冲洗,加入山羊抗鼠或山羊抗兔二抗(1∶2000),室温孵育2 h,暗室曝光,扫描图像,以Quantity One 4.4.0凝胶定量软件分析目的蛋白相对表达量。
6.免疫荧光法:将经高压灭菌处理的玻片置入6孔板中,取对数生长中期SGC7901/MDR细胞悬液,以2×105/孔接种于6孔板内玻片上,连续培养24 h,以埃索美拉唑(0、100μg/mL)预处理 24 h 后取出玻片,PBS漂洗3次,冷丙酮固定,山羊封闭血清室温封闭1 h,加入V-H+-ATPases一抗(1∶175)或P-gp一抗(1∶75),以PBS代替一抗作为阴性对照,4℃孵育过夜。加入荧光二抗(1∶150)室温孵育 1 h,加入 DAPI(2μg/mL)染核,1 h 内 DSY5000i倒置荧光显微镜(北京长恒荣创科技有限公司)下观察、拍照。
三、统计学分析
结 果
一、PPIs抑制 SGC7901/MDR细胞内 V-H+-ATPases和 P-gp、MRP1表达
与对照组(0μg/mL)相比,20μg/mL 埃索美拉唑预处理24 h可显著抑制SGC7901/MDR细胞内的 V-H+-ATPases、P-gp 表达(P <0.05),浓度升至50μg/mL 可显著抑制 MRP1表达(P <0.05),抑制作用随药物浓度的升高而增强,呈浓度依赖性(P<0.05)(见图1A)。然而不同浓度埃索美拉唑预处理对 SGC7901细胞内的 V-H+-ATPases、P-gp、MRP1表达均无明显影响(见图1B)。
与对照组相比,10μg/mL泮托拉唑预处理24 h可显著抑制SGC7901/MDR细胞内的V-H+-ATPases表达(P <0.05),浓度升至 20μg/mL 可显著抑制P-gp、MRP1表达,抑制作用随药物浓度的升高而增强,呈浓度依赖性(P<0.05)(见图1C)。
经100μg/mL埃索美拉唑预处理 24 h的SGC7901/MDR细胞,免疫荧光法检测显示胞内VH+-ATPases、P-gp表达和定位与对照组相比有明显改变(见图2)。
二、PPIs抑制SGC7901/MDR细胞内PI3K/Akt/mTOR/HIF-1α信号通路
与对照组相比,20μg/mL埃索美拉唑预处理24 h可显著抑制SGC7901/MDR细胞内的PI3K、HIF-1α 表达(P <0.05),浓度升至 50μg/mL 可显著抑制Akt、mTOR 表达(P <0.05),抑制作用随药物浓度的升高而增强,呈浓度依赖性(P<0.05)(见图3A)。然而100μg/mL埃索美拉唑预处理才能显著抑制SGC7901细胞内的PI3K表达(P<0.05),不同浓度埃索美拉唑预处理对Akt、mTOR、HIF-1α表达均无明显影响(见图3B)。
与对照组相比,50μg/mL泮托拉唑预处理24 h可显著抑制 SGC7901/MDR细胞内的 PI3K、Akt、mTOR、HIF-1α 表达(P <0.05),抑制作用随药物浓度的升高而增强,呈浓度依赖性(P<0.05)(见图3 C)。
三、PPIs对TSC1/2-Rheb信号通路以及Akt底物和TSC2磷酸化的影响
TSC1/2-Rheb信号通路为PI3K/Akt/mTOR信号旁路。与对照组相比,50μg/mL埃索美拉唑预处理24 h可显著抑制SGC7901/MDR细胞内的TSC1、TSC2、Rheb表达(P <0.05),抑制作用随药物浓度的升高而增强,呈浓度依赖性(P<0.05)(见图4A)。然而80μg/mL埃索美拉唑预处理才能显著抑制SGC7901细胞内的 TSC2表达(P<0.05),且浓度达 100μg/mL时TSC2表达不再继续降低;100μg/mL埃索美拉唑预处理才能显著抑制TSC1表达(P<0.05);不同浓度埃索美拉唑预处理对Rheb表达均无明显影响(见图4B)。
Akt底物的磷酸化水平可直接反映Akt磷酸化调节的作用强度,埃索美拉唑和泮托拉唑预处理24 h均可呈浓度依赖性地抑制SGC7901/MDR细胞内的Akt底物磷酸化;Ser939、Thr1462是 Akt磷酸化TSC2的两个重要位点,埃索美拉唑和泮托拉唑预处理24 h均能在一定程度上抑制这两个位点的磷酸化(见图5)。

图1 PPIs预处理对SGC7901/MDR、SGC7901细胞内V-H+-ATPases和P-gp、MRP1表达的影响

图2 埃索美拉唑预处理对SGC7901/MDR细胞内V-H+-ATPases、P-gp表达和定位的影响(×200)

图3 PPIs预处理对SGC7901/MDR、SGC7901细胞内PI3K/Akt/mTOR/HIF-1α信号通路的影响

图4 PPIs预处理对SGC7901/MDR、SGC7901细胞内TSC1/2-Rheb信号通路的影响
四、PPIs对SGC7901/MDR细胞内TSC1/2复合物表达的影响呈时间依赖性
TSC1、TSC2为抑癌基因,然而PPIs可呈浓度依赖性地抑制SGC7901/MDR细胞内的TSC1/2复合物表达,为此本实验进一步在未缓冲培养体系中以80μg/mL埃索美拉唑分别预处理SGC7901/MDR细胞 15 min 和3 h、6 h、12 h、18 h、24 h,结果显示埃索美拉唑对胞内TSC1/2复合物表达的影响呈时间依赖性,处理 3 h后 TSC1、TSC2表达较对照组(15 min)显著升高(P<0.05),18 h后与对照组相比无明显差异,24 h后较对照组显著降低(P<0.05);TSC1表达于处理12 h后达峰值,TSC2表达处理6 h后即达峰值(见图6)。

图5 PPIs预处理对SGC7901/MDR细胞内Akt底物和TSC2磷酸化的影响
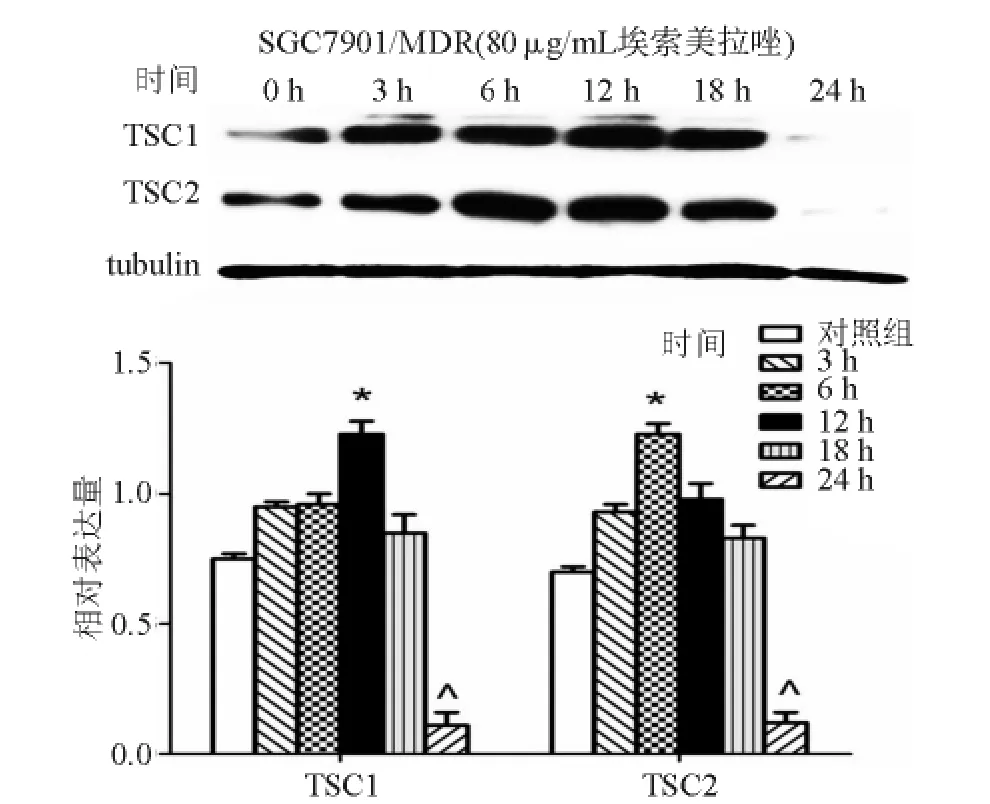
图6 埃索美拉唑预处理对SGC7901/MDR细胞内TSC1/2复合物表达的影响
五、V-H+-ATPases siRNA抑制 SGC7901/MDR细胞内PI3K/Akt/mTOR/HIF-1α信号通路和P-gp表达
V-H+-ATPases siRNA干扰可明显抑制SGC7901/MDR细胞内的 V-H+-ATPases表达(见图 7A),PI3K、Akt、TSC1、TSC2、mTOR、HIF-1α、P-gp 表达随之显著下调(P<0.05)(见图7B),与PPIs预处理结果相似。
六、雷帕霉素阻断mTOR抑制SGC7901/MDR细胞内 HIF-1α、P-gp表达
与对照组相比,20μg/mL雷帕霉素处理24 h可显著抑制SGC7901/MDR细胞内的mTOR表达(P<0.05),抑制作用随药物浓度的升高而增强,呈浓度依赖性(P <0.05),浓度升至80μg/mL 时,mTOR表达几乎完全被抑制;HIF-1α、P-gp表达随mTOR表达的下降同步降低(P<0.05)(见图8)。

图7 V-H+-ATPases siRNA对SGC7901/MDR细胞内PI3K/Akt/mTOR/HIF-1α信号通路和 P-gp表达的影响

图8 雷帕霉素阻断mTOR对SGC7901/MDR细胞内HIF-1α、P-gp表达的影响
讨 论
肿瘤细胞的MDR是一个复杂的多因素现象,与其局部微环境如缺氧、酸化、炎症以及肿瘤细胞本身密切相关[12]。为阐明肿瘤发生的生物学机制,寻找更好的治疗途径,近年来肿瘤细胞代谢及其微环境重新成为研究热点。肿瘤细胞处于一种缺氧、酸化的微环境中,细胞内外存在pH梯度,可激活细胞内部一系列信号转导分子,导致化疗耐药[13]。空泡型质子泵对肿瘤细胞内外pH梯度的维持起关键调节作用,PPIs则可抑制空泡型质子泵活性,逆转肿瘤细胞内外pH梯度,导致胞内pH值偏酸性,胞外pH值偏碱性,并增强胃癌细胞的化疗敏感性[8]。本课题组的前期实验发现PPIs增强胃癌细胞化疗敏感性的作用与抑制 MDR蛋白P-gp、MRP1表达有关。本实验证实PPIs可呈浓度依赖性地抑制SGC7901/MDR细胞内的空泡型质子泵表达,同时抑制P-gp、MRP1表达,对SGC7901细胞内的上述蛋白表达则无明显影响,证实PPIs可通过抑制空泡型质子泵下调MDR蛋白表达并改变其胞内定位,从而提高耐药胃癌细胞的化疗敏感性。
PI3K/Akt信号通路可调节细胞生长、增殖,促进细胞骨架形成。TSC1、TSC2为抑癌基因,位于Akt下游、mTOR上游,是PI3K/Akt信号通路的重要调控位点。这两种基因产物形成的复合物TSC1/2对Rheb(一种大量存在于脑组织中的Ras超家族GTP结合蛋白)具有GAP(GTP酶活化蛋白)活性,是mTOR的关键负性调节因子。Akt激活后磷酸化TSC2的Ser939和Thr1462两个位点,导致TSC1/2复合物解离,对Rheb的抑制作用解除,从而活化下游 mTOR 分子,发挥生物学效应[14~16]。mTOR 不仅是PI3K/Akt信号通路的下游分子,亦为HIF-1α的上游关键调节分子[10,11]。
本实验发现PPIs可呈浓度依赖性地抑制SGC7901/MDR细胞内的PI3K、Akt及其下游分子mTOR、HIF-1α表达以及Akt底物磷酸化,表明PPIs可抑制耐药胃癌细胞内的PI3K/Akt/mTOR/HIF-1α信号通路,然而本实验未能进一步检测PI3K/Akt信号通路中其他中间信号分子的表达情况以及Akt自身的磷酸化状态。后续研究拟完善上述不足,并在应用PPIs的同时或其后上调PI3K/Akt信号通路,如HIF-1α和MDR蛋白表达不受影响,则更能说明PPIs的作用依赖于该通路。此外,本实验还发现PPIs引起的PI3K/Akt信号通路抑制使TSC2磷酸化水平降低,导致TSC1/TSC2复合物表达及其GAP活性在一定时间段内上调,从而抑制Rheb的表达和活性及其下游mTOR分子。表明PPIs还可抑制PI3K/Akt/mTOR信号旁路,通过抑制TSC1/2-Rheb信号通路以抑制其下游mTOR分子而发挥作用。
PPIs对TSC1/2复合物表达的影响呈时间依赖性,提示PI3K/Akt/mTOR信号通路的调控可能存在某种负反馈回路。已有研究证实PI3K/Akt信号通路的下游组分如mTOR可通过抑制胰岛素受体底物蛋白(IRS)功能而阻断该信号通路的进一步活化[17]。由此推测mTOR表达抑制可能反馈性地促进该信号通路活化,进而抑制TSC1/2复合物表达,具体机制有待进一步研究。
空泡型质子泵高表达于肿瘤细胞的细胞膜和胞内酸性囊泡膜,正常组织细胞膜不表达空泡型质子泵,其主要功能为将肿瘤细胞胞质中糖酵解产生的H+泵出胞外或泵入胞内酸性囊泡,以维持胞质内弱碱性或偏中性的环境,此环境与肿瘤细胞对化疗药物耐药密切相关。本研究以V-H+-ATPases siRNA抑制SGC7901/MDR细胞内的V-H+-ATPases表达后,PI3K/Akt/mTOR/HIF-1α信号通路及其旁路分子表达均受抑,MDR蛋白P-gp表达下调,证实了空泡型质子泵在胃癌细胞化疗耐药机制中的重要地位,表明PPIs系通过抑制空泡型质子泵,逆转耐药胃癌细胞的胞内外pH梯度而发挥作用。
mTOR对天然化合物雷帕霉素非常敏感,雷帕霉素对mTOR的抑制作用有极强的特异性[14]。本实验发现雷帕霉素可呈浓度依赖性地抑制mTOR表达,下游HIF-1α、P-gp表达同步降低,雷帕霉素浓度升至80μg/mL时,mTOR表达几乎完全被抑制,HIF-1α、P-gp表达亦如是,证实两者位于mTOR分子下游,表明PPIs系通过抑制PI3K/Akt/mTOR信号通路,进而抑制HIF-1α和MDR蛋白表达以增强耐药胃癌细胞的化疗敏感性。有研究[18]发现HIF-1α可通过调节p53和NF-κB决定胃癌细胞的化疗敏感性,抑制HIF-1α可使胃癌细胞对5-氟尿嘧啶和顺铂的敏感性显著增强。表明抑制PI3K/Akt/mTOR信号通路可通过不同途径逆转胃癌细胞对化疗药物的 MDR,包括调节 p53、NF-κB以及下调MDR蛋白等。关于HIF-1α能否通过MDR蛋白参与肿瘤细胞的MDR及其在非缺氧条件下的耐药机制,均有待进一步研究。
综上所述,PPIs可通过抑制耐药胃癌细胞的胞内空泡型质子泵逆转肿瘤细胞内外pH梯度,使肿瘤细胞胞内酸化、胞外碱化,从而改变肿瘤细胞微环境,同时抑制胞内PI3K/Akt/mTOR信号通路,进而抑制HIF-1α和MDR蛋白表达,以增强肿瘤细胞的化疗敏感性,逆转其对化疗药物的MDR。PPIs有望成为一种新型肿瘤化疗佐剂,但目前研究尚处于细胞和动物水平,相关临床试验仍有待开展。
1 Cunningham D,Allum WH,Stenning SP;MAGIC Trial Participants.Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer[J].N Engl J Med,2006,355(1):11-20.
2 Liang XJ,Aszalos A.Multidrug transporters as drug targets[J].Curr Drug Targets,2006,7(8):911-921.
3 Aller SG,Yu J,Ward A,et al.Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding[J].Science,2009,323(5922):1718-1722.
4 Fais S.Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism[J].J Intern Med,2010,267(5):515-525.
5 Fais S,De Milito A,You H,et al.Targeting vacuolar H+-ATPases as a new strategy against cancer[J].Cancer Res,2007,67(22):10627-10630.
6 Huber V,De Milito A,Harguindey S,et al.Proton dynamics in cancer[J].J Transl Med,2010,8:57.
7 Luciani F,Spada M,De Milito A,et al.Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs[J].J Natl Cancer Inst,2004,96(22):1702-1713.
8 Chen M,Zou X,Luo H,et al.Effects and mechanisms of proton pump inhibitors as a novel chemosensitizer on human gastric adenocarcinoma(SGC7901)cells[J].Cell Biol Int,2009,33(9):1008-1019.
9 Rafiee P,Theriot ME,Nelson VM,et al.Human esophageal microvascular endothelial cells respond to acidic pH stressby PI3K/AKT and p38 MAPK-regulated induction of Hsp70 and Hsp27[J].Am J Physiol Cell Physiol,2006,291(5):C931-C945.
10 Emerling BM,Akcakanat A.Targeting PI3K/mTOR signaling in cancer[J].Cancer Res,2011,71(24):7351-7359.
11 Wouters BG,Koritzinsky M.Hypoxia signalling through mTOR and the unfolded protein response in cancer[J].Nat Rev Cancer,2008,8(11):851-864.
12 Morin PJ.Drug resistance and the microenvironment:nature and nurture[J].Drug Resist Updat,2003,6(4):169-172.
13 DeClerck K,Elble RC.The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy[J].Front Biosci,2010,15:213-225.
14 Huang J,Manning BD.The TSC1-TSC2 complex:a molecular switchboard controlling cell growth[J].Biochem J,2008,412(2):179-190.
15 Manning BD,Tee AR,Logsdon MN,et al.Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway[J].Mol Cell,2002,10(1):151-162.
16 Aspuria PJ,Tamanoi F.The Rheb family of GTP-binding proteins[J].Cell Signal,2004,16(10):1105-1112.
17 Manning BD.Balancing Akt with S6K:implications for both metabolic diseases and tumorigenesis[J].J Cell Biol,2004,167(3):399-403.
18 Rohwer N,Dame C,Haugstetter A,et al.Hypoxiainducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappaB[J].PLoS One,2010,5(8):e12038.
猜你喜欢
甘肃科技(2020年21期)2020-04-13 00:34:18
中华胃食管反流病电子杂志(2017年3期)2017-01-16 01:23:38
中国卫生标准管理(2015年17期)2016-01-20 09:26:46
中国卫生标准管理(2015年14期)2016-01-15 02:58:35
中国当代医药(2015年24期)2015-03-01 02:06:09
中国药业(2014年17期)2014-05-26 09:07:55
中国药业(2014年19期)2014-05-17 03:12:19
河南医学研究(2014年4期)2014-02-27 14:52:28
