
线粒体DNA突变合并肾脏损害
2012-05-10 13:08谢红浪许书添何群鹏郭锦洲桂兰兰陈惠萍刘志红
肾脏病与透析肾移植杂志 2012年6期
谢红浪 许书添 何群鹏 杨 柳 郭锦洲 桂兰兰 陈惠萍 刘志红
线粒体病是指因遗传基因缺陷引起的线粒体结构和功能异常导致的一组多系统细胞呼吸链及能量代谢异常的疾病。其中线粒体DNA(mtDNA)缺失或点突变使编码线粒体氧化代谢过程必需的酶或载体发生障碍,线粒体不能利用糖原和脂肪酸等底物产生足够的三磷酸腺苷(ATP)供细胞使用,从而导致能量代谢障碍。根据病变部位,通常将其分为线粒体肌病、线粒体脑肌病和线粒体脑病三类。
近年,随着对线粒体基因结构和功能的深入研究发现,线粒体病几乎可导致全身各个器官和系统功能损害(如肌肉、中枢神经系统、心脏、消化、内分泌、眼等),线粒体病合并肾脏损害的报道也逐渐增多[1-3]。本文报告3例合并肾脏损害的mtDNA点突变的患者,以期提高临床医师对此类疾病的认识和重视。
对象和方法
研究对象 2011年5月至2012年9月在南京军区南京总医院全军肾脏病研究所住院、相互间无血源关系的3例患者,均经临床诊断线粒体病,分析其个人史和家庭史,肾脏损害的临床特征、肾外多系统受累的表现,治疗及预后,并分析肾脏病理特点。
所有患者及例2之父、母、弟均空腹留取EDTA抗凝血标本,分离外周血白细胞检测mtDNA基因突变;3例患者同时送肾组织标本检测mtDNA基因突变情况。
基因组标本准备 采用TIANamp Genomic DNA Kit提取外周血白细胞基因组DNA,DNA浓度>100 ng/μl,体积﹥ 40 μl;纯度均应达到:260/280 >1.8,260/230>1.2。
引物准备 将引物从原始的100p稀释为10p(100p的原始引物10 μl+去离子水90 μl)。
PCR扩增
(1)将去离子水、PCR缓冲液、dNTP和Mg+溶液提前融化(注意:PCR缓冲液、dNTP和Mg+溶液必须完全融化才能使用)。
(2)体系:含有 dH2O 17.3 μl,10 × Buffer(含Mg+)2.5 μl,dNTP(2.5 mM)2 μl,引物(10p)2 μl,TAKARA 热启动 TaqE(5U/μl)0.2 μl,DNA 1.0 μl,总体积为 25 μl。
(3)PCR程序:变性开始阶段为95.0℃,15 min,扩增阶段为 95.0℃,30s;退火 58.0℃,30s,延伸72.0℃,30s,最终延伸 72.0℃,10 min,共扩增36圈。
(4)基因扩增所用的引物见表1。

表1 基因扩增引物
测序分析 根据碱基序列判断检测结果的是否存在mtDNA基因缺失或突变改变。
结 果
一般情况 3例患者中,男性2例,女性1例,年龄14~30岁。3例患者均无高血压,体型偏瘦,例1和例2的体质量指数(BMI)分别为13.3 kg/m2和13.5 kg/m2(表2)。

表2 3例患者的一般情况
个人史和家族史 例1生长和第二性征发育较同龄人迟缓伴智力障碍,例2有类固醇性糖尿病,例3为早产儿。例1及例2之母有糖尿病史,例2同时有尿毒症家族史(表3)。
肾脏损害 发现肾脏病病程在7月内。所有患者均以水肿和尿量减少起病,伴胱抑素C增高,2例合并低白蛋白血症及高脂血症。尿液检查均无高血压和镜下血尿,但肾小管损伤明显尿 N-乙酰-β-D-氨基葡萄糖苷酶和视黄醇结合蛋白升高(表4)。

表3 3例患者的个人史及家族史
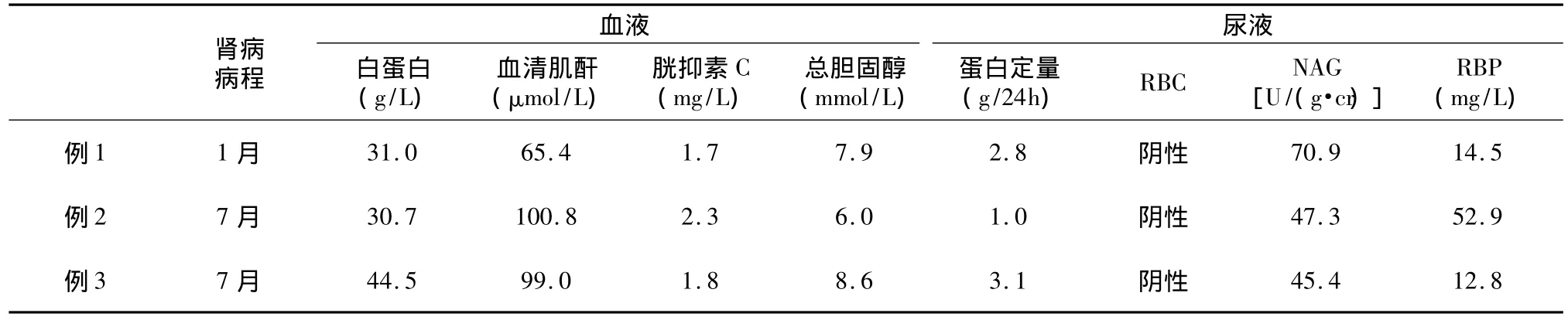
表4 3例患者肾脏损害的特点
肾活检组织病理 例1免疫荧光IgM++,呈颗粒状弥漫沉积于系膜区及血管袢;光镜示局灶节段肾小球硬化性病变(1/2肾小球废弃)(图1A)。例2免疫荧光IgM++,C3+,呈细颗粒状弥漫沉积于系膜区及血管袢;光镜示肾小球轻度系膜增生性病变但存在肾小动脉节段透明变性。2例患者肾组织IV型胶原α3、α5均正常。例3外院肾穿刺免疫荧光IgM+,光镜示局灶节段性肾小球硬化性病变。
电镜:例1肾小球节段袢塌陷,足细胞胞质及肾小管上皮细胞胞质内见大小不一、形态不规则的线粒体(图1B、C)。例2肾小球轻度系膜增生性病变,足细胞和肾小管上皮细胞胞质见肿胀变形的线粒体,见双核足细胞(图1D)。例3无电镜结果。
肾外损害 2例患者符合糖尿病诊断标准,糖化血红蛋白明显升高(7.8%~10.4%),2例有高乳酸血症(>2.0 mmol/L)。神经系统损害:癫痫发作(1例),脑梗塞(1例),听力丧失(1例),高频听力下降(2例),智力障碍(2例),视野缺失(1例)等。例1有心律失常(表5)。
基因诊断 3例患者均经基因测序证实存在mtDNA 3243 A>G突变位点,例2之母和弟弟也检出与患者相同突变位点。例1和例2同时送检肾活检组织,未检出相同突变位点。
治疗及预后 例1和例2激素治疗无效,经对症支持治疗后,尿蛋白未转阴。例3曾用激素+环磷酰胺冲击治疗无效,后佐以左卡尼汀、艾地苯醌、辅酶Q10等治疗,蛋白尿部分缓解,血清肌酐稳定(表5)。
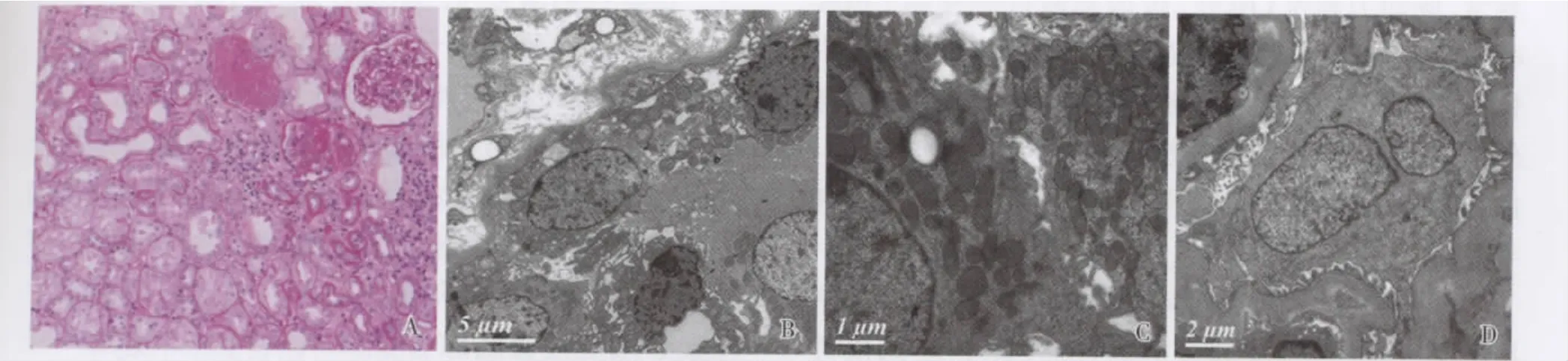
图1 线粒体病的肾组织活检病理

表5 3例患者肾外损害的表现和预后
讨 论
线粒体病的病因和发病机制 近10年来线粒体生物发展迅速。现已知人类线粒体DNA(mtDNA)约为16.6kb,编码37个基因,包括22种tRNA,2种 rRNA和13种与呼吸链相关的蛋白质[2]。原发性mtDNA异常包括点突变、缺失或重复等。点突变符合母系遗传特点,mtDNA发生突变的概率比细胞核DNA(nDNA)高6~17倍,且不同细胞间的mtDNA突变量相差很大,具有异质性。当mtDNA突变量达到一定阈值时,即可影响细胞功能导致临床疾病,小剂量突变则不一定致病。
线粒体病几乎可影响全身各个器官,临床表现多种多样。骨骼肌常表现为肌病、肌张力减退和运动能力减弱;中枢神经系统呼吸暂停、低张性麻痹、嗜睡、运动神经元病、共济失调、卒中样发作、偏瘫、痉挛、癫痫、痴呆、脑白质营养不良、肌阵挛、皮质盲、偏头痛、多发性神经病(感染或运动)及神经元性膀胱等。感音性耳聋和心脏疾病发生率也很高(心肌病、心律失常及传导阻滞等),内分泌异常引起糖尿病、甲状旁腺功能减退、甲减、低肾素低醛固酮血症和生长激素不足等。消化系统功能受损出现肝功能异常、肠动力紊乱(呕吐、腹泻和假性肠梗阻)和吸收不良。血液系统损害表现铁粒幼细胞性贫血、中性粒细胞减少和血小板减少。眼部症状包括进行性外眼肌麻痹、眼肌麻痹、视网膜色素变性、上睑下垂、白内障、视神经萎缩及失明。此外,还可累及皮肤和毛发。
线粒体病合并肾脏疾病的报道多见于儿童,常出现Fanconi综合征或肾病综合征、肾小管-间质性疾病、Bartter综合征和肾小管性酸中毒等。成人线粒体病与儿童不同,女性患者多见,多遵循母系遗传特点;肾活检以局灶节段性肾小球硬化(FSGS)最常见[3],肾小管-间质性疾病和多囊性疾病其次。儿童散发病例多见,女性不占优势,且FSGS的发生率低于肾小管-间质疾病[4]。
编码tRNALEU基因的3243位点A突变为G是最经典和最常见的mtDNA基因异常,该点突变最初在儿童MELAS综合征患者(肌病、脑肌病、乳酸酸中毒和卒中样发作)中证实,进一步研究发现,糖尿病人群中该突变发生率高达1%,更常见糖尿病、耳聋、胃肠道和/或神经肌肉症状。1997年一项大型研究发现,糖尿病患者具母系遗传特点和(或)感音性耳聋者,发生蛋白尿性肾脏疾病和终末期肾功能衰竭的比例特别高[5]。Emma 等[6]对 Alport患者重新筛查,从90例中发现了2例3243 A>G突变者。在一项研究报道的27个3243 A>G突变病例中,2/3(20/27)为女性,先证者之母或其他家庭成员多有糖尿病和(或)耳聋,确诊年龄14~50岁。肾脏病理多表现为FSGS(22/27),4例为慢性肾小管-间质性肾炎,1例为多囊性肾脏疾病;其中2例见特殊的小动脉透明变性和肌细胞坏死样病变。电镜观察通常无特殊发现,偶见足细胞或肾小管上皮细胞中集聚大量形态多样的线粒体及双核或多核的足细胞。
泛醌或辅酶Q10(CoQ10)是参与线粒体呼吸链电子穿梭的重要成分[7],其编码基因突变是另一种最常见mtDNA突变性疾病。Emma等[6]已在一些激素抵抗型肾病综合征和脑肌病的儿童患者证实存在CoQ10合成缺陷和COQ2基因突变,COQ2基因主要编码对羟苯甲酸聚戊烯转移酶,参与CoQ10醌基合成。COQ2基因突变还见于一些遗传性或早发性激素抵抗性肾病综合征,COQ1-PDSS2基因[8]和COQ6基因[9]突变也可导致类似肾脏损害。此类疾病早期诊断尤为重要,因为是可治的一种线粒体缺陷,采用CoQ10[5~10 mg/(kg·d)]治疗可改善临床预后。
线粒体病的诊断 当患者具以下临床特征时,应考虑线粒病[4,10]:(1)不明原因的多个需氧器官受累,如糖尿病、神经-肌肉病变、肥厚性心肌病和肾脏损害(肾病综合征、Fanconi综合征等);(2)具母系遗传性家族性糖尿病;(3)伴感音性耳聋的糖尿病。
疑诊线粒体病时,可先行静息状态下血清乳酸检测,必要时检测血液丙酮酸、肌酶、脑脊液乳酸、尿氨基酸和有机酸等,但这些并非线粒体病所特有,如运动后和危重患者均可出现血乳酸升高;近端小管功能损伤者可因尿液丢失增多而致血乳酸正常。骨骼肌活检可见“破碎红纤维”,但由于线粒体病的异质性,肌肉活检也可正常,故应尽量选择临床功能受累的器官(如心肌)。评估呼吸链组分和功能的免疫组化、组化和分光光度计检测方法,也有助线粒体病的诊断,但个体间差异较大。可取肾活检组织送检呼吸链复合物,骨骼肌送检CoQ10,皮肤活检获取成纤维细胞经培养后行CoQ10生物合成率生化检测。
采用PCR检测mtDNA点突变或Southern Blotting检测基因缺失,是诊断线粒体病的金标准,但需鉴定突变基因的致病性,且可能遗漏nDNA突变。送检标本选择至关重要,最常送检的标本为血液和肌肉,有作者报道从尿沉渣细胞中可检出mtDNA 突变[11]。Hotta等[1]报道1 例伴严重肾功能衰竭的患者,其尿沉渣mtDNA突变的程度低,而患者之子虽无肾脏损害证据,却从尿沉渣中检出极高比例的mtDNA突变细胞。因此Hotta等[1]认为从尿沉渣中检出mtDNA突变细胞并不能预测患者是否发病及疾病的严重程度。
国外已有大量线粒体病的研究(http:∥mitomap.org)及肾脏损害的报道,国内也早已认识到线粒病可继发肾脏损害[12,13],但相关研究和病例报道较少[14,15]。Levinger等[16]报道,在出现临床症状的患者中已证实150多种mtDNA的致病性点突变和100多种mtDNA缺失。本组3例患者均证实3243 A>G突变。
本研究发现肾活检病理并非能确诊线粒体病肾脏损害,因为线粒体病的组织学改变无特征性,必需除外其他疾病继发的FSGS。Hotta等[1]报道,其他原因继发性FSGS(如肾发育不良、反流性肾病和肥胖相关性肾病等)的肾小球体积变异较大(8~16×106μm3,平均 12 ×106μm3),而原发性 FSGS 的肾小球体积(3~6×106μm3,平均5×106μm3)明显大于线粒体病-FSGS者(2.5~4×106μm3,平均3.5×106μm3),三组间统计学差异十分显著。电镜观察线粒形态和数量异常也不具特异性,但双核或多核足细胞可能有一定的诊断价值。Moulonguet等[3]报道,小动脉透明变性有提示诊断的意义,可能与小动脉肌细胞坏死有关,并导致FSGS病变。
本研究的不足之处为仅检测部分患者家族成员mtDNA突变情况,且受检测方法限制,未能检测出突变与未突变细胞的比例。
总之,本文首次在国内报道了3例伴肾脏损害的线粒体病患者,提示这类患者并非罕见,临床医师应加强对此类疾病的认识。
1 Hotta O,Inoue CN,Miyabayashi S,et al.Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR)gene mutation.Kidney Int,2001,59(4):1236 -1243.
2 Emma F,MontiniG,SalviatiL,etal.Renalmitochondrial cytopathies.Int J Nephrol,2011,2011:609213.
3 Moulonguet LD,HillGS,Chedin P,etal.Focalsegmental glomerulosclerosis associated with mitochondrial cytopathy.Kidney Int,2000,58(5):1851 -1858.
4 Hall AM,Unwin RJ,Hanna MG,Duchen MR.Renal function and mitochondrial cytopathy(MC):more questions than answers?QJM,2008,101(10):755 -766.
5 Jansen JJ,Maassen JA,van der Woude FJ,et al.Mutation in mitochondrial tRNA(Leu(UUR))gene associated with progressive kidney disease.J Am Soc Nephrol,1997,8(7):1118 -1124.
6 Emma F,BertiniE,SalviatiL,etal.Renalinvolvementin mitochondrial cytopathies.Pediatr Nephrol,2012,27(4):539 -550.
7 Quinzii CM,Hirano M.Coenzyme Q and mitochondrial disease.Dev Disabil Res Rev,2010,16(2):183 -188.
8 López LC,Schuelke M,Quinzii CM,et al.Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2(PDSS2)mutations.Am J Hum Genet,2006,79(6):1125-1129.
9 Heeringa SF,Chernin G,Chaki M,et al.COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness.J Clin Invest,2011,121(5):2013 -2024.
10 Guéry B,Choukroun G,No?l LH,et al.The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu)gene mutation.J Am Soc Nephrol.2003,14(8):2099 -2108.
11 Löwik MM,Hol FA,Steenbergen EJ,et al.Mitochondrial tRNALeu(UUR)mutation in apatientwith steroid-resistantnephrotic syndrome and focalsegmentalglomerulosclerosis.NephrolDial Transplant,2005,20(2):336 - 341.
12 刘志红.局灶节段性肾小球硬化的诊断:摆脱组织形态学束缚的努力.肾脏病与透析肾移植杂志,2009,18(1):49-51.
13 刘志红.微小病变性肾病及局灶节段性肾小球硬化//黎磊石,刘志红.中国肾脏病学.北京:人民军医出出版社,2008:364-386.
14 全军肾脏病研究所学术委员会,肾病综合征、糖尿病、癫痫、预激综合征和神经肌源性损害.肾脏病与透析肾移植杂志,2012,21(3):295-298.
15 黄 倩,曾彩虹,刘志红.IgA肾病合并线粒体病.肾脏病与透析肾移植杂志,2012,21(3)291 -294.
16 Levinger L,MorlM,Florentz C.MitochondrialtRNA 30 end metabolism andhuman disease.Nucleic Acids Res,2004,32(18):5430-5441.
猜你喜欢
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
中外医疗(2016年15期)2016-12-01
中外医疗(2015年11期)2016-01-04
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
西南军医(2015年6期)2015-01-23
癌变·畸变·突变(2014年1期)2014-03-01
河南医学研究(2014年1期)2014-02-27
