
PVP/FF复合纳米纤维的制备与表征
2012-03-25 11:18袁彩艳聂华丽朱利民
合成纤维工业 2012年6期
袁彩艳,权 静,聂华丽,2* ,朱利民
(1.东华大学化学化工与生物工程学院,上海201620;2.东华大学 纺织面料技术教育部重点实验室,上海201620)
静电纺丝技术是指聚合物溶液或熔体被喷射拉伸成纤维的技术,可制备出直径为纳米级的超细纤维,最小直径可至1 nm。与传统的制备聚合物纤维的方法相比,静电纺丝法制备的超细纤维膜具有比表面积大和孔隙率高、质轻等优点,在组织工程、药物和催化剂载体、伤口敷料、过滤、传感器、模板、防护织物、纳米电子元件等众多领域具有潜在的应用价值[1-3]。目前对聚合物溶液或熔体通过静电纺丝技术制备纳米纤维的例子非常多,但有关小分子化合物的研究还很少见。
二苯丙氨酸(FF)是形成阿尔茨海默氏症和帕金森综合症等疾病有关的淀粉样多肽的核心结构单元。由于FF具有氢键、β折叠结构以及芳香环间的π-π作用,具有良好的自组装性质,因此能在水或有机溶剂中通过分子之间的氢键、范德华力、π-π相互作用、配位键等非共价键作用而自组装形成稳定的类似蛋白质的二级或三级结构[4];另外,由于 FF良好的可降解性、生物相容性等优点使其备受关注。自从FF被发现以来,不论是在医学还是纳米科技领域都引起了人们很大的研究兴趣。一直以来,许多科学家探索用不同的方法来调控FF自组装过程[4-6]。但由于FF单体相对分子质量小,溶液浓度低等特性,纯FF单体难以静电纺丝。目前也没有关于FF与其他聚合物的混合物溶液进行静电纺丝的报道。聚乙烯基吡咯烷酮(PVP)是一种水溶性的高分子聚合物,具有良好的的生物降解性和生物相容性,而且PVP可纺性较好。由于其分子中同时含有亲水基团和亲油基团,因此可与多种物质,尤其是含羧基、羟基、胺基及其他含活性氢的化合物形成络合物,在许多领域都具有广泛的应用[7-8]。因此,作者将FF与PVP的混合溶液进行静电纺丝,为进一步研究FF自组装行为打下基础。
1 实验
1.1 原料与试剂
FF:纯度95%,上海毕得医药有限公司产;PVP:K60,重均分子量为3.6×105,上海运宏化工有限公司产;二甲基乙酰胺(DMAc)、甲醇:分析纯,国药集团上海化学试剂公司产。
1.2 设备
静电纺丝装置采用削平的5号不锈钢注射针头(内径0.5 mm)作为喷射细流的毛细管,连接上海苏特电器有限公司生产的ZGF2000型高压发生器,由美国Cole-Parmer公司生产的微量注射泵控制纺丝液流量,纤维采用铝箔平板接受。
1.3 PVP/FF复合纳米纤维的制备
将适量的FF和PVP溶解在甲醇/DMAc体积比为85/15的溶液中,制备PVP质量分数为10%,FF 质量分数依次为 0,0.4%,0.6%,2%,5%的共混纺丝液。在16 kV的静电压,接受板离喷丝口距离为25 cm,环境温度为(18±1)℃,环境相对湿度为(60±2)%,纺丝液流速分别为0.1,0.2,0.4,0.8,1.0 mL/h 的条件下静电纺丝。制得的纤维试样见表1。

表1 PVP/FF复合纳米纤维的制备条件及其平均直径Tab.1 Preparation conditions for PVP/FF nanofibers and their average diameters
1.4 测试与表征
表面形貌:采用日本 JEOL公司的 JSM-5600LV扫描电子显微镜(SEM)观察纤维的表面形貌。从纤维的SEM图片中随机选取100根纤维,用Image J软件测量其直径,计算平均直径。
X射线衍射(XRD)分析:采用日本RigaKu公司的D/Max-BR X射线衍射仪进行XRD分析。分别取不同试样10 mg,在5°~ 60°下,在40 mV,300 mA条件下进行XRD分析。
热稳定性分析:采用德国耐驰仪器制造有限公司的TG 209 F1型热重分析仪测定纤维的热稳定性。称取5~10 mg复合纳米纤维于坩埚中,在氮气气氛下,以10℃/min的速率进行升温,温度为0~600℃,得到热失重(TG)分析曲线。
全反射红外光谱(ATR-FTIR)分析:取2 mg纤维试样,采用美国Thermo Fisher公司的Nicolet Nexus 670型傅里叶变换红外光谱仪在700~3 500 cm-1内扫描,测定纤维的红外光谱。
2 结果与讨论
2.1 FF含量对纤维形态的影响
由图1可见:F1~F4试样结构均匀,表面光滑,没有出现交联和串珠现象;而F5形貌较差,粗细不均匀、表面粘连现象较为明显。由表1看出,随着复合纤维中FF含量的增加,纤维直径逐渐变小,当FF的质量分数增加至5%时,纤维直径也相应增加。这是由于随着复合纤维中FF含量的增加,纤维中PVP的相对含量变小,从而导致溶液黏度变小,导致纤维直径下降。当FF的含量增加到一定程度,溶液黏度太小,虽然能够形成纤维,但是在喷射过程中,有液滴喷射到纤维膜的表面,导致还没有完全干燥的纤维发生粘连、串珠甚至断裂;另一种原因是由溶剂的挥发造成的,即在喷射过程中,由于溶剂没有完全挥发直接将液滴喷射到已经干燥的纤维表面,从而将其溶解,导致电镜下观察到的纤维出现粘连、串珠现象。

图1 PVP/FF复合纳米纤维的SEM照片Fig.1 SEM images of PVP/FF nanofibers
2.2 纺丝液流速对纤维形态的影响
由图2可见:当纺丝液流速为0.1~1.0 mL/h时,都可以得到纳米纤维,F5~F9试样的形态较好,粗细均匀,表面光滑,没有颗粒附着。但F5试样形貌较差,出现了粘连现象。由表1表明,随着纺丝液流速的增加,纤维平均直径也逐渐增大。这是由于纺丝液的流速增大,导致喷丝口形成的液滴初始体积变大,从而导致纤维直径增大。如图2e所示,当流速为1.0 mL/h时,得到的纤维直径分布不均匀且有粘连现象发生。当流速为0.1 mL/h时,时常会造成喷丝口堵塞。这是由于流速过慢,溶剂挥发,使得喷丝口纺丝液浓度增加,从而容易造成喷丝口堵塞。因此,流速在0.2~0.6 mL/h时静电纺丝效果最佳。

图2 不同流速下制备的纳米纤维的SEM照片Fig.2 SEM images of nanofibers prepared at different flow rate
2.3 纤维中各组分的物理状态及相互作用
2.3.1 XRD 分析
由图 3 可见:FF 在 2θ为 10.60°,14.22°,21.24°等处出现特征衍射峰;随着PVP/FF复合纳米纤维中FF含量的增加,PVP/FF复合纳米纤维的FF晶体衍射峰基本消失,并且PVP的两个无定型态特征“驼峰”强度也降低(F5试样)。这说明PVP/FF复合纳米纤维中FF与PVP发生复合作用,失去了原有的晶体结构,处于一种高度分散的无定形态[9]。

图3 FF和PVP及其复合纳米纤维的XRD光谱Fig.3 XRD patterns of FF and PVP and their composite nanofibers
2.3.2 TG 分析
从图4可以看出:FF在170℃左右开始缓慢分解,快速分解温度分为180~200℃与290~340℃两个阶段;PVP的分解温度集中在380~440℃,随着FF含量的增加,复合材料的分解温度逐渐向低温方向移动;同时,随着FF含量的增加,复合纳米纤维的分解温度范围也变得更宽;复合纳米纤维的分解温度为200~450℃。这说明组分FF与PVP之间存在着某种相互作用,这种作用使得PVP的稳定性减弱,分解温度降低,这与XRD分析结果一致。
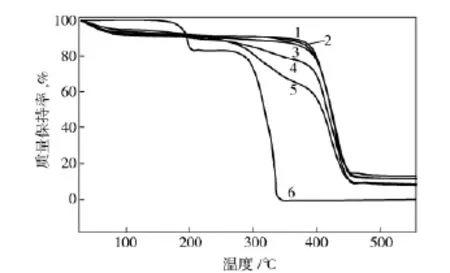
图4 FF和PVP及其复合纳米纤维的TG曲线Fig.4 TG curves of FF and PVP and their composite nanofibers
2.3.3 红外光谱分析
由图5可见:由于FF分子是两个苯丙氨酸的缩合,因此FF的红外吸收光谱在1 734 cm-1和1 669 cm-1这两个位置上出现C=O特征峰,在3 369 cm-1的吸收峰为—NH—的特征吸收峰,2 893,2 958 cm-1为羧酸氢键的伸缩振动吸收峰,1 533,1471 cm-1等处为苯环的特征吸收峰。
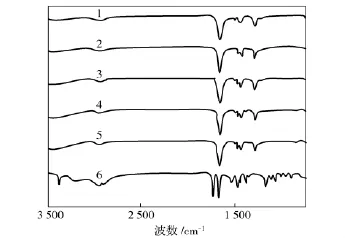
图5 FF和PVP及其复合纳米纤维的红外光谱Fig.5 FTIR spectra of FF and PVP and their composite nanofibers
在PVP/FF复合纳米纤维膜的图谱上,随着PVP/FF复合纳米纤维中FF含量的增加,在3 369 cm-1处—NH—的特征吸收峰变弱或消失。此外,在1 533,1 471 cm-1处的FF的苯环伸缩振动峰也从复合纳米纤维膜的图谱上消失;另外,FF和PVP分别在1 734,1 669,1 659 cm-1处有 C=O的伸缩振动吸收峰,而在PVP/FF复合纳米纤维膜的图谱上则融合成一个峰并向低波数方向移动到1 658 cm-1。这些现象说明FF分子与PVP之间通过氢键发生相互作用,具有较好的相容性。
3 结论
a.随着FF含量的增加,PVP/FF复合纳米纤维直径先减小后增加,当FF质量分数小于5%时,溶液的可纺性较好。
b.随着流速的增大,PVP/FF复合纳米纤维直径有逐渐增大趋势。流速为0.2~0.6 mL/h时得到的复合纳米纤维分布均匀性较好,形貌较佳。
c.PVP/FF复合纳米纤维中FF与PVP发生复合作用,失去了原有的晶体结构,处于一种高度分散的无定形态。
d.随着FF含量的增加,PVP/FF复合纳米纤维的分解温度逐渐向低温方向移动。同时,PVP/FF复合纳米纤维的分解温度范围也变得更宽。
e.在PVP/FF复合纳米纤维中FF与PVP发生氢键作用,均匀地分散到纳米纤维中,具有良好的相容性。
[1] Greiner A,Wendorff J H.Electrospinning:A fascinating method for the preparation of ultrathin fibers[J].Angew Chem Int Edit,2007,46(30):5670 -5703.
[2] Huang Zhengming,Zhang Yanzhong,Kotaki M,et al.A review on polymer nanofibers by electrospinning and their applications in nanocomposites[J].Compos Sci Technol,2003,63(15):2223-2253.
[3] Huang Zhangming,Zhang Yanzhong.Micro-structures and mechanical performance of co-axial nanofibers with drug and protein cores and polycaprolactone shells[J].Chem J Chinese Univ,2005,26(5):968 -972.
[4] Reches M,Gazit E.Casting metal nanowires with in discrete self-assembled peptide nanotubes[J].Science,2003,300(5619):625-627.
[5] Singh G,Bittner A M,Loscher S,et al.Electrospinning of diphenylalanine nanotubes[J].Adv Mater,2008,20(12):2332-2336.
[6] Kim J,Han T H,Kim Y I,et al.Role of water in directing diphenylalanine assembly into nanotubes and nanowires[J].Adv Mater,2010,22(5):583 -587.
[7] Esnaashari S,Javadzadeh Y,Batchelor H K,et al.The use of microviscometry to study polymer dissolution from solid dispersion drug delivery systems[J].Int J Pharm,2005,292(1/2):227-230.
[8] Rawlinson C F,Williams A C,Timmins P,et al.Polymer-mediated disruption of drug crystallinity[J].Int J Pharm,2007,336(1):42-48.
[9] 余灯广,申夏夏,张晓飞,等.速溶电纺载药PVP纳米纤维膜制备与表征[J].高分子学报,2009(11):1170-1174.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
小哥白尼(趣味科学)(2022年2期)2022-05-25
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
云南化工(2021年7期)2021-12-21
孩子(2020年11期)2020-11-17
大众科学(2020年7期)2020-10-26
趣味(数学)(2019年6期)2019-10-17
小天使·六年级语数英综合(2018年1期)2018-10-08
科学大众·小诺贝尔(2016年11期)2017-01-10
燕山大学学报(2015年4期)2015-12-25
