
Red同源重组敲除nagE和manX对大肠杆菌发酵生产氨基葡萄糖的影响
2012-02-09 09:38:46陈欣刘龙李江华刘杰堵国成陈坚
生物工程学报 2012年3期
陈欣,刘龙,李江华,刘杰,堵国成,陈坚
1 糖化学与生物技术教育部重点实验室,江苏 无锡 214122
2 江南大学生物工程学院,江苏 无锡 214122
3 江苏江山制药有限公司,江苏 靖江 214500
4 江南大学粮食发酵工艺与技术国家工程实验室,江苏 无锡 214122
氨基葡萄糖 (Glucosamine,2-amino-2-deoxy-D-glucose,GlcN) 又称氨基葡糖或葡糖胺,是葡萄糖的一个羟基被氨基取代后的化合物[1-4],在保健食品和医药领域具有广泛应用,如能特异性地作用于关节软骨,有效治疗风湿性关节炎;可抑制白血病细胞K562的生长;可参与肝肾解毒,发挥抗炎、护肝的作用等[5-7]。在日本和美国,GlcN作为食品配料广泛应用。但令人遗憾的是,目前国内上市的GlcN产品还比较少,相关产品的开发空间和潜力巨大。GlcN的生产方法可分为3种:甲壳素水解法、生物转化法[8-9]和微生物发酵法[1-4]。甲壳素水解是目前生产GlcN的主要方法,但原料来源受限、环境污染严重、部分消费者服用后会过敏;生物转化法生产的酶的价格高、生产成本高,转化时间长,难于实现工业化生产[1-4,10-11];因此微生物发酵生产GlcN吸引了越来越多研究者的关注[12-14]。相对于水解法和生物转化法,微生物发酵法具有以下优点:1) 发酵时间较短,生产强度较高;2) 原料来源不受地域和季节限制,产品无鱼腥味;3) 对环境污染小;4) 产品来自微生物,消费者服用后不会出现过敏。但目前国内外关于 GlcN发酵生产的研究寥寥无几。
对大肠杆菌中 GlcN合成代谢途径的研究[1,4,15-16]表明:进入细胞内的葡萄糖在磷酸葡萄糖异构酶 (PGI) 的催化下转化成6-磷酸葡萄糖,后者在氨基葡萄糖合成酶 (GlmS) 催化下生成GlcN,氨基葡萄糖乙酰化酶 (Gna1) 催化 GlcN转化成乙酰氨基葡萄糖 (GlcNAc) (图1)。本研究室通过在大肠杆菌 Escherichia coli ATCC 25947中过量表达氨基葡萄糖合成酶 (由基因glmS编码) 和氨基葡萄糖乙酰化酶 (由基因gna1编码),成功构建了一株可高效合成氨糖的重组大肠杆菌E. coli-glms-gna1。通过乙酰化将GlcN转化为GlcNAc可显著降低GlcN对细胞生长和代谢的抑制作用,在下游纯化过程中用弱酸处理即可将GlcNAc脱乙酰获得最终产品GlcN,因此,GlcN和GlcNAc都是本文中的目标发酵产物。但前期的研究发现[4],由于 GlcN依靠甘露糖磷酸转移系统 (Mannose PTS,ⅡMan,由manXYZ操纵子编码) 和葡萄糖磷酸转移系统(glucose PTS,ⅡGlc,由pstG基因编码) 对其进行磷酸化后从胞外向胞内转运,GlcNAc依靠ⅡMan和乙酰氨基葡萄糖磷酸转移系统 (GlcNAc PTS,ⅡNAG,由基因nagE编码) 对其进行磷酸化后从胞外向胞内转运,导致氨糖产量降低。因此,要想在胞外积累高浓度的GlcN或GlcNAc,就必须减弱或阻断GlcN或GlcNAc从胞外到胞内的转运。manXYZ操纵子由X、Y、Z三个基因构成,其中manX基因编码甘露糖特异性磷酸转移复合酶的组分ⅡA和ⅡB蛋白,manX基因的缺失对甘露糖特异性磷酸转移复合酶活性影响较大,通过敲除nagE和manX基因,有望阻止胞外GlcN或GlcNAc向胞内的转运,进而增加产量[17-18]。
近年来发展起来的依赖λ噬菌体的Red重组系统,可以直接利用含同源臂的PCR线性片段,替换出同源臂内的目的基因。该系统利用λ噬菌体3个蛋白质Exo、Bet和Gam,Cam抑制大肠杆菌的RecBCD核酸外切酶V的活性,使外源线性DNA不会立即被降解,Exo和Bet引导线性片段与同源区发生重组置换,而且效率较传统的同源重组方法高几十倍[19-20]。我们从基因突变菌株中通过PCR反应扩增出突变基因及其两翼 DNA片段,利用该系统替代 E. coli-glms-gna1中相应基因,构建了 nagE及manX缺失的大肠杆菌工程菌株,使其丧失氨基葡萄糖磷酸转移功能,以此增加胞外氨糖产量。
1 材料与方法
1.1 试剂与材料
1.1.1 菌株和质粒
E. coli-glms-gna1:本实验保藏,将来自大肠杆菌 E. coli基因组的氨基葡萄糖合成酶基因(glmS) 和氨基葡萄糖乙酰化酶基因 (gna1) 通过pET-28 (a) 在E. coli ATCC 25947 (DE3) 中进行了克隆和表达而获得;E. coli JW0665-1 (F-,∆(araD-araB) 567, ∆lacZ4787 (::rrnB-3),∆nagE728::kan,lambda-,rph-1,∆ (rhaD-rhaB) 568,hsdR514);E. coli JW1806-1 (F-,∆(araD-araB) 567,ΔlacZ4787 (::rrnB-3),lambda-,ΔmanX741::kan,rph-1,∆ (rhaD-rhaB) 568,hsdR514),用于提供被卡那霉素抗性基因 (Kanr)插入失活的nagE及manX片段,购自美国耶鲁大学菌种保藏中心;质粒pKD46和pCP20购自美国耶鲁大学菌种保藏中心。
1.1.2 酶和试剂
胰蛋白胨和酵母粉购自 Oxoid公司;LA DNA聚合酶和 PCR产物回收试剂盒购自TaKaRa公司 (大连);卡那霉素、氨苄青霉素、引物合成、L-阿拉伯糖和质粒提取试剂盒购于Sangon生工生物工程 (上海) 有限公司;其余试剂均为国产分析纯。
1.1.3 仪器
电转化仪 (GenePulser Xcell),Bio-Rad公司;台式高速离心机,日本Hitech公司;DYY-6C型电泳仪,北京六一仪器厂;凝胶成像系统,Bio-Rad公司;PCR仪,Bio-Rad公司;7 L发酵罐 (NEW BRUNSWICK SCIENTIFIC,G107-09)。
1.2 方法
1.2.1 培养基
LB培养基:酵母膏5 g,蛋白胨10 g,NaCl 10 g,琼脂20 g,定容至1 L,pH 7.0。
发酵培养基:葡萄糖10.0% (W/V),蛋白胨1.2% (W/V),酵母膏 2.4% (W/V),甘油 0.4% (V/V),KH2PO417 mmol/L,K2HPO472 mmol/L。
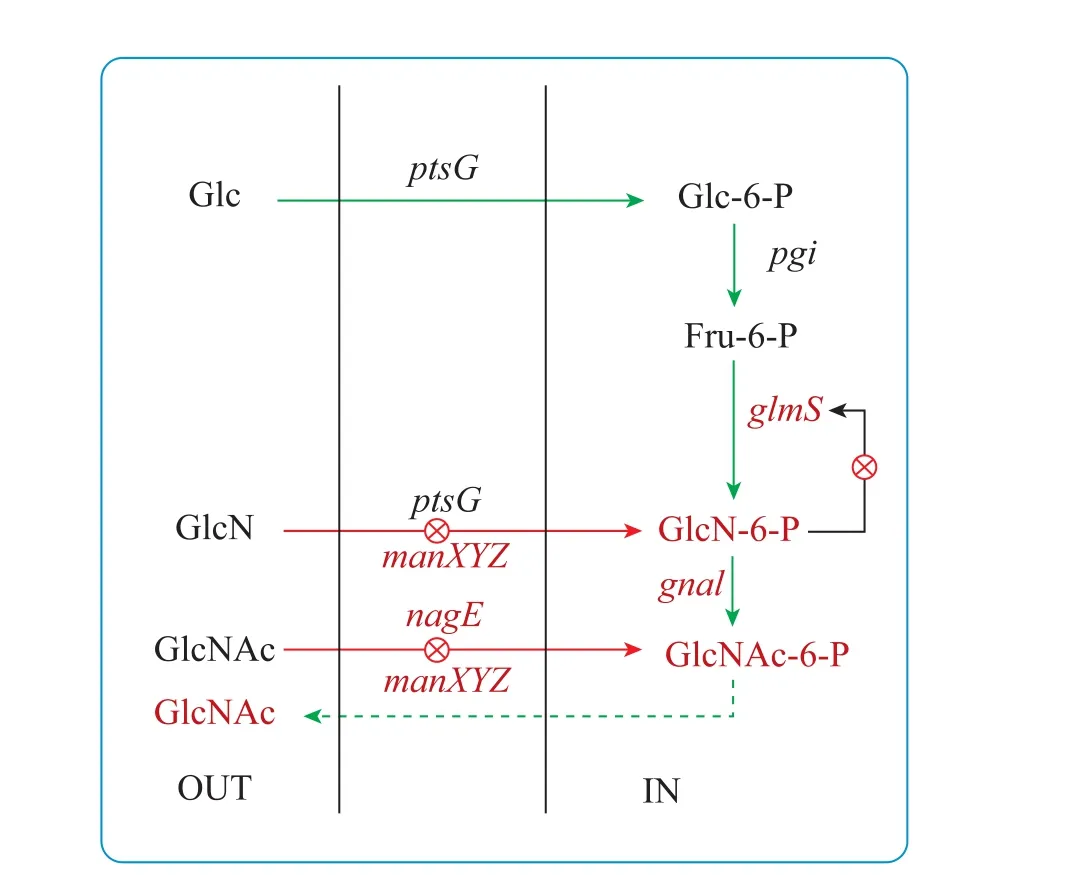
图1 大肠杆菌中氨基葡萄糖合成途经Fig. 1 Biosynthetic pathways of glucosamine in E. coli. Glc: glucose; Fru-6-P: fructose-6-P; ptsG: glucose transporter gene; GlcN-6-P: glucosamine-6-P; GlcNAc-6-P: N-acetylglucosamine-6-P; glmS: glucosamine synthase; gna1: glucosamine-6-phosphate acetyltransferase; nagE: GlcNAc-specific transporter gene; manXYZ: mannose transporter gene; OUT: outside the cell; IN: inside the cell.
1.2.2 引物设计
根据E. coli JW0665 nagE基因上下游800 bp设计引物NU和ND,扩增得到3.5 kb的突变序列;根据E. coli JW1806-1 manX基因上下游500 bp序列设计引物MU和MD,扩增得到2.3 kb的突变序列,同时根据kanr内部序列设计鉴定引物K1和K2,用于对转化子的验证 (表1)。
1.2.3 感受态细胞制备
从30 ℃培养16~20 h的平板中挑取一个单菌落,转接到含20 mL LB培养基的250 mL三角瓶中,于 30 ℃下 200 r/min培养过夜,按0.5%的接种量转接含有50 mL LB培养基的500 mL三角瓶中,于30 ℃、200 r/min培养至OD600≈0.6,将三角瓶置于冰中预冷 30 min,于超净台内将培养液转移到预冷的 50 mL无菌离心管中,冰上放置10 min;4 ℃、4 000 r/min离心10 min;弃上清,加入少量预冷的无菌水重悬菌体,4 ℃、4 000 r/min离心10 min;重复1次无菌水洗菌体;最后用预冷的 10%甘油洗涤 1次,4 ℃、4 000 r/min离心10 min,弃上清,将菌体悬浮于0.8 mL 10%的甘油中,按每管50 µL感受态细胞进行分装,或保存于−80 ℃备用。
制备用于同源重组的 E. coli JW0665-1 (pKD46) 或E. coli JW1605-1 (pKD46) 感受态细胞时,当菌液OD600≈0.2时加入1 mmol/L的L-阿拉伯糖诱导Red重组酶表达,30 ℃、200 r/min培养至菌液最终OD600≈0.6,其他操作同上。
1.2.4 电转化
取出感受态细胞,在冰浴中融化;加入待转化的1~2 µL DNA或1 µL pKD46或pCP20,用粗口吸头轻柔混合;将上述混合物转入预冷的0.2 cm电转杯中,冰浴中预冷10 min;打开电转仪,将参数设定为2 500 V,25 mF,200 Ω,电击时间约为5 ms;从冰中取出电转杯,用纸巾吸去表面的水分,放入样品槽中;电击后立即加入1 mL LB培养基,30 ℃、200 r/min培养1.5 h后将转化后的感受态细胞涂布在含有抗生素的LB平板上于30 ℃培养进行筛选。扩增片段转化含有 pKD46的感受态细胞时,电转化后的培养条件为30 ℃培养1.5 h,然后升温至42 ℃培养12~16 h以去除pKD46;利用pCP20消除转化子中kanr时,电转化后的培养条件为30 ℃培养2 h,然后升温至42 ℃培养 12~16 h以去除pCP20。
1.2.5 转化子验证
将电转化后长出的转化子分别用 NU/ND、 MU/MD及鉴定引物K1/K2进行菌落PCR验证(表2),考察nagE和manX基因是否已被kanr基因替换。
1.2.6 发酵实验
在7 L发酵罐中配制发酵培养基,装液量为4 L,接入E. coli-glms-gna1、E. coli-glms-gna1-∆nagE和E. coli-glms-gna1-∆nagE-∆manX,接种后37 ℃、400 r/min培养,通气量240 L/h,至OD600达到0.6 (约4 h),加入乳糖 (其在发酵罐中的浓度为5 g/L),诱导重组质粒pET28 (a)-glms-gna1中glmS和gna1基因表达,继续发酵至20 h。于接种后6 h、8 h、10 h、12 h、14 h、16 h、18 h、20 h进行生物量、GlcN和GlcNAc产量的分析。
1.2.7 生物量、GlcN和GlcNAc的检测方法
生物量采用干重法测定。GlcN的检测参考Elson-Morgan法[4,21]:取发酵液5.0 mL加入5 mL离心管中,8 000 r/min离心8 min,取0.5 mL离
心上清液加入乙酰丙酮试剂1.0 mL,90 ℃水浴处理1 h,冷却至室温,慢慢加入96% (V/V) 乙醇10 mL,然后加入DMAB试剂1.0 mL,混合均匀。混合后室温放置1 h,530 nm处比色,根据标准曲线计算GlcN产量。GlcNAc用HPLC方法检测[22],Agilent 1200,RID检测器,Alltech C18柱(250×4.6 mm,5 μm),流动相:70%乙腈,流速0.7 mL/min,柱温30 ℃,进样体积为10 μL。
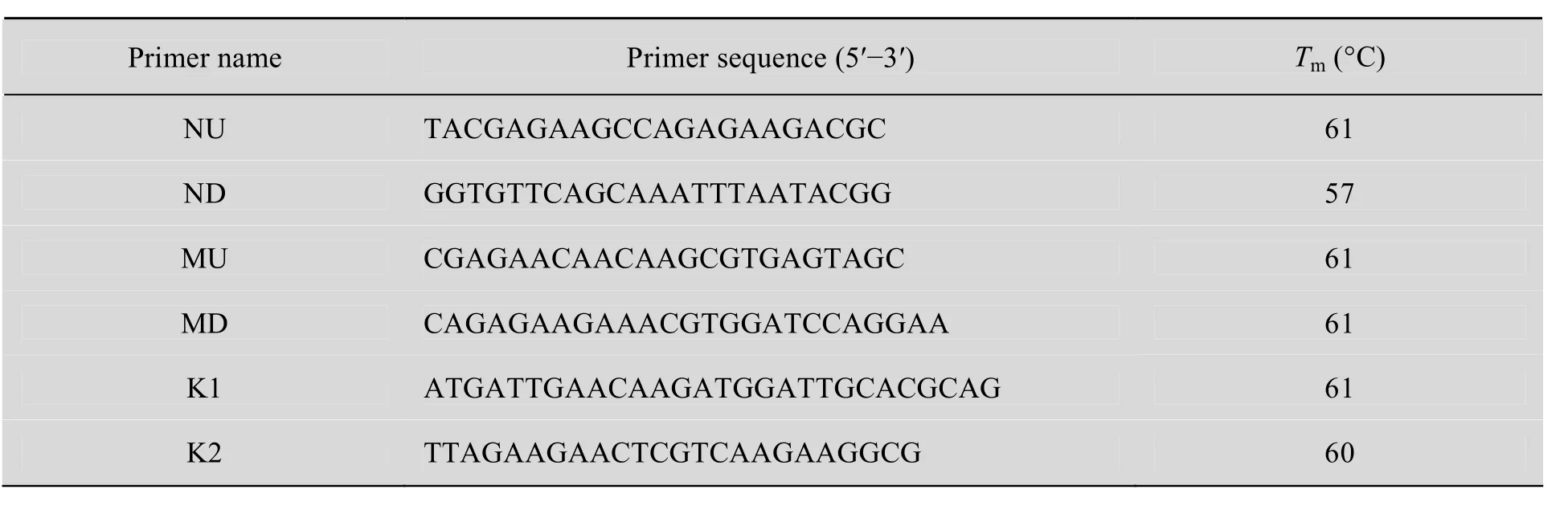
表1 本实验中使用的PCR系列引物Table 1 PCR primers used in this work
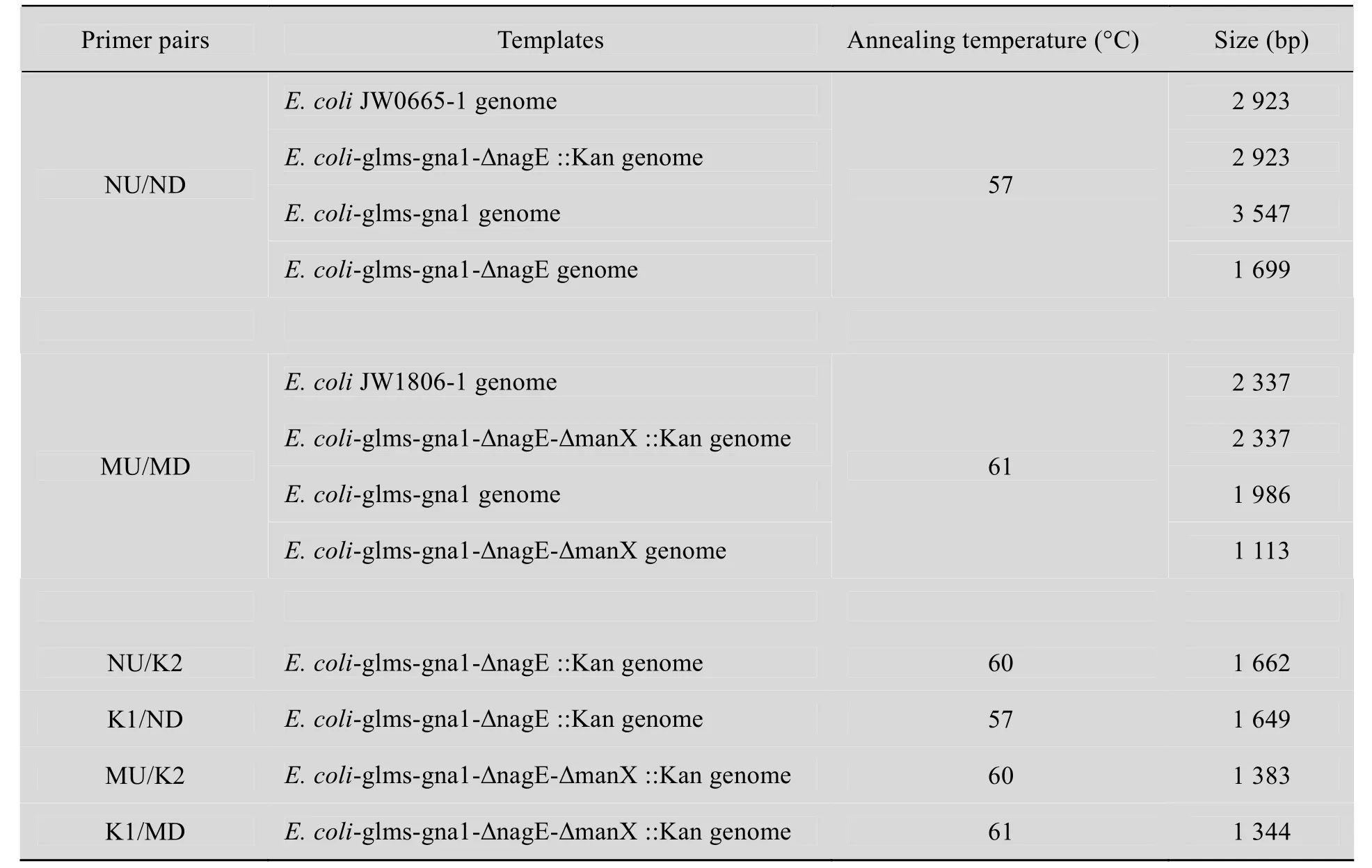
表2 转化子菌落PCR反应中使用的引物、模板、退火温度及产物大小Table 2 Primer pairs, templates and annealing temperature used for the transformant colony PCR and the size of PCR products
2 结果与分析
2.1 卡那抗性基因替代nagE及manX基因
利用引物NU/ND从E. coli JW0665-1中扩增出的 DNA片段,通过 Red重组替代 E. coliglms-gna1基因组nagE基因,转化子验证结果如图2所示。泳道2为E. coli-glms-gna1- ∆nagE::kan菌落PCR产物,大小为2 923 bp,泳道3为原菌株E. coli-glms-gna1菌落PCR产物,大小为3 547 bp,利用鉴定引物NU/K2和K1/ND分别对 E. coli-glms-gna1、E. coli JW0665-1和 E. coli-glms- gna1-∆nagE::Kan进行菌落PCR,泳带4和8为对照,无扩增产物,泳道5、6、7中条带大小均为1 649 bp,泳道9、10中条带大小均为1 662 bp,说明携带kanr基因的PCR扩增片段已经替代了nagE基因,得到nagE基因插入失活的菌株E. coli-glms-gna1- ∆nagE::kan。
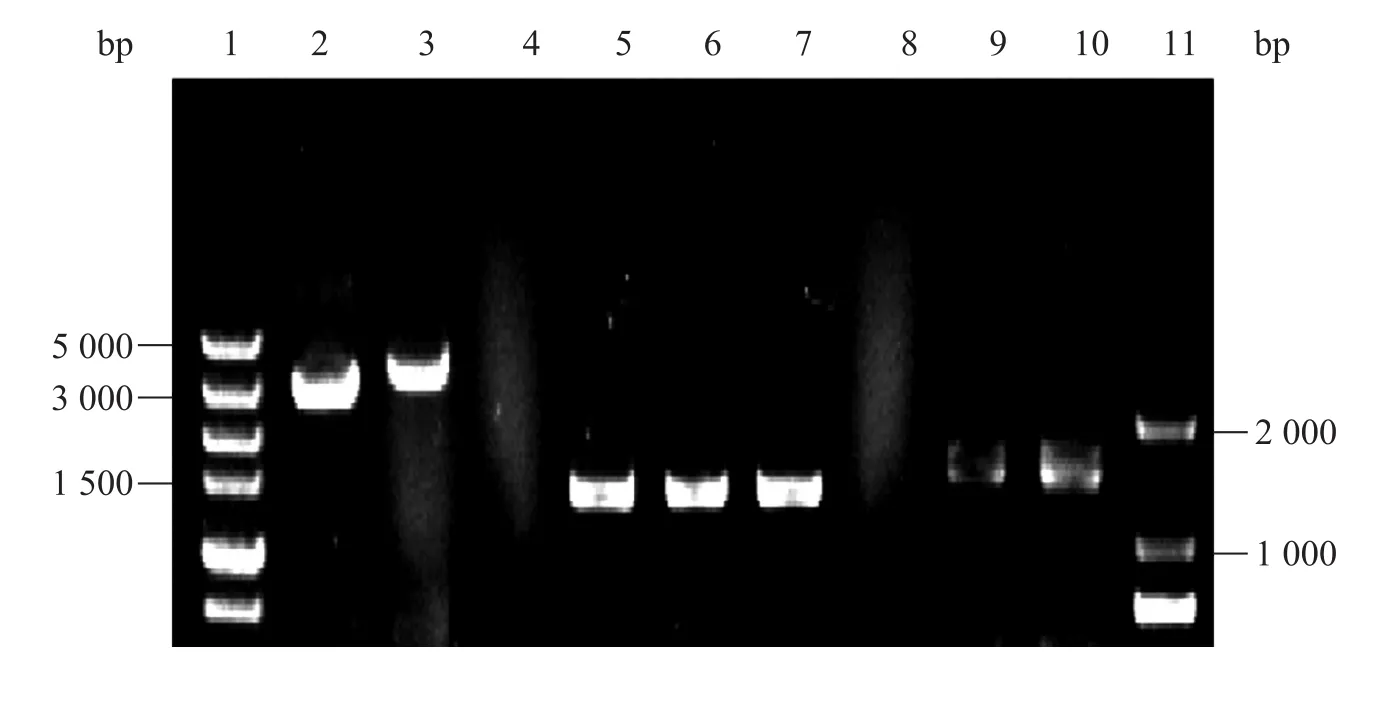
图2 E. coli-glms-gna1-∆nagE::kan菌落PCR电泳结果Fig. 2 Colony PCR products of E. coli-glms-gna1-∆nagE::kan. 1: DL 5 000 DNA marker; 2: colony PCR products of E. coli-glms-gna1-∆nagE::kan amplified by primers NU and ND (2 923 bp); 3: colony PCR products of E. coli-glms-gna1 amplified by primers NU and ND (3 547 bp); 4: colony PCR products of E. coli-glms-gna1 amplified by primers K1 and ND; 5: colony PCR products of E. coli JW0665-1 amplified by primers K1 and ND (1 649 bp); 6, 7: colony PCR products of E. coli-glms-gna1-∆nagE::kan amplified by primers K1 and ND (1 649 bp); 8: colony PCR products of E. coli-glms-gna1 amplified by primers NU and K2; 9: colony PCR products of E. coli JW0665-1 amplified by primers NU and K2 (1 662 bp); 10: colony PCR products of E. coli-glms-gna1- ∆nagE::kan amplified by primers NU and K2 (1 662 bp); 11: DL 2000 DNA marker.
利用引物 MU/MD从 E. coli JW1806-1中扩增出的 DNA片段,通过 Red重组替代E. coli-glms-gna1-∆nagE基因组manX基因。转化子验证结果如图3所示,泳道1和2为E. coli-glms-gna1-∆nagE菌落PCR产物,大小为2 923 bp,泳道3、4分别为菌株E. coli JW1806-1和 E. coli-glms-gna1-∆nagE-∆manX::Kan菌落PCR产物,大小为3 547 bp,利用鉴定引物NU/K2和K1/ND分别对E. coli-glms-gna1-∆nagE、E. coli JW1806-1和E. coli-glms-gna1-∆nagE-manX::Kan进行菌落PCR,泳道5和8为对照,无扩增产物,泳道6、7条带大小均为1 344 bp,泳道9、10条带大小均为 1 383 bp,说明携带 kanr基因的PCR扩增片段已经替代了manX基因。
2.2 卡那抗性基因的去除
将质粒 pCP20转化 E. coli-glms-gna1-∆nagE::kan,去除kanr,验证结果如图4所示。泳道1、2为E. coli-glms-gna1-∆nagE菌落PCR产物,大小为1 699 bp,泳道3、4分别为E. coli JW0665-1和 E. coli-glms-gna1-∆nagE::kan菌落PCR产物,大小为2 923 bp,泳道5为E. coli-glmsgna1落PCR产物,大小为3 547 bp,说明kanr已经从转化子中去除。
将质粒pCP20转化E. coli-glms-gna1-∆nagE-∆manX::kan,去除kanr,得到E. coli-glms-gna1-∆nagE-∆manX,验证结果如图5所示。泳道1、2为E. coli-glms-gna1-∆nagE-∆manX菌落PCR产物,大小为 1 113 bp,泳道 3、4分别为 E. coli-glms-gna1-∆nagE::kan和E. coli JW1806-1菌落PCR产物,大小为2 337 bp,说明kanr已经从转化子中去除。

图3 E. coli-glms-gna1-∆nagE-∆manX::kan菌落PCR电泳结果Fig. 3 Colony PCR products electrophoresis analysis result of E. coli-glms-gna1-∆nagE-∆manX::kan. 1, 2: colony PCR products of E. coli-glms-gna1-∆nagE amplified by primers MU and MD (1 986 bp); 3: colony PCR products of E. coli JW1806-1 amplified by primers MU and MD (2 337 bp); 4: colony PCR products of E. coli-glms-gna1-∆nagE-∆manX::kan amplified by primers MU and MD (2 337 bp); 5: colony PCR products of E. coli-glms-gna1 amplified by primers K1 and MD; 6: colony PCR products of E. coli JW1806-1 amplified by primers K1 and MD (1 344 bp); 7: colony PCR products of E. coli-glms-gna1-∆nagE-∆manX::kan amplified by primers K1 and MD (1 344 bp); 8: colony PCR products of E. coli-glms-gna1 amplified by primers MU and K2; 9: colony PCR products of E. coli JW1806-1 amplified by primers MU and K2 (1 383 bp); 10: colony PCR products of E. coli-glms-gna1-∆nagE-∆manX::kan amplified by primers MU and K2 (1 383 bp); 11: DL 10 000 DNA marker.
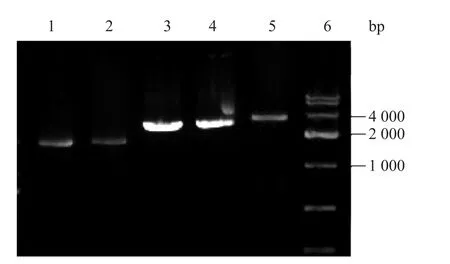
图4 E. coli-glms-gna1-∆nagE菌落PCR产物电泳结果Fig. 4 Colony PCR products electrophoresis analysis result of E. coli-glms-gna1-∆nagE. 1, 2: colony PCR products of E. coli-glms-gna1-∆nagE amplified by primers NU and ND (1 699 bp); 3: colony PCR products of E. coli JW0665-1 amplified by primers NU and ND (2 923 bp); 4: colony PCR products of E. coli-glms-gna1- ∆nagE::kan amplified by primers NU and ND (2 923 bp); 5: colony PCR products of E. coli-glms-gna1 amplified by primers NU and ND (3 547 bp); 6: DL 10 000 DNA marker.
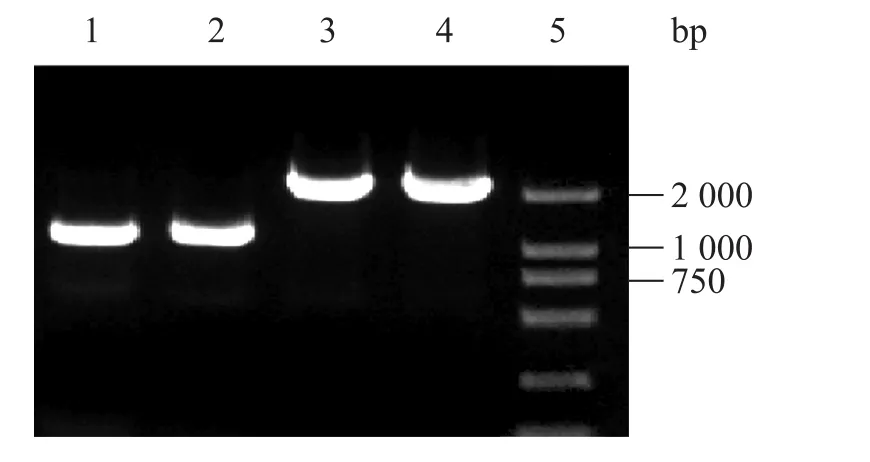
图5 E. coli-glms-gna1-∆nagE-∆manX菌落PCR产物电泳结果Fig. 5 Colony PCR products electrophoresis analysis result of E. coli-glms-gna1-∆nagE-∆manX. 1, 2: colony PCR products of E. coli-glms-gna1-∆nagE-∆manX amplified by primers MU and MD (1 113 bp); 3: colony PCR products of E. coli-glms-gna1- nagE-∆manX::kan amplified by primers MU and MD (2 337 bp); 4: colony PCR products of E. coli JW1806-1 amplified by primers MU and MD (2 337 bp); 5: DL 2 000 DNA marker.
2.3 发酵实验结果
2.3.1 E. coli-glms-gna1-∆nagE发酵结果
从图 6和表 3可以看出,E. coli-glmsgna1-∆nagE和对照菌株E. coli-glms-gna1的菌浓在 14 h时均达到最高值,分别为 5.42 g/L和4.68 g/L,说明nagE基因的敲除对与细胞膜合成相关的物质的代谢未造成较大影响,细胞用于合成细胞膜的前体物质GlcNAc可通过manX等基因编码产物将GlcN转运到胞内而得到补偿,这可以从发酵前期12 h内E. coli-glms-gna1-∆nagE的比生长速率较对照菌株高看出。
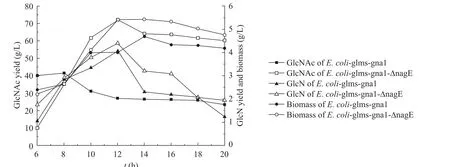
图6 E. coli-glms-gna1-∆nagE发酵实验结果Fig. 6 Results of GlcN and GlcNAc production by E. coli-glms-gna1-∆nagE.
在发酵过程中,就GlcN产量而言,E. coliglms-gna1-∆nagE在12 h达到最大值4.38 g/L,随后呈现明显下降趋势,这可能是因为nagE基因的敲除主要抑制GlcNAc向胞内的转运,细胞生长对碳源的需求导致GlcN能依靠ptsG编码的酶ⅡCBGlc而进入细胞,而对照菌株在 12 h时GlcN产量达到最高值 4.06 g/L,E. coli-glmsgna1-∆nagE的GlcN产量高于对照菌株,这一方面可能由于nagE基因的敲除减少了GlcNAc向胞内的转运,另一方面可能是由于菌浓的不同造成的。通过计算单位菌体的GlcN得率发现,对照菌株在 12 h时,单位菌体的 GlcN产量为1.02 g/g DCW,高于此时E. coli-glms-gna1-∆nagE的0.81 g/g DCW。对照菌株的GlcN生产强度在8 h时达到最高值0.35 g/(L·h),而E. coli-glmsgna1-∆nagE的GlcN生产强度在10 h时达到最高值0.38 g/(L·h),随后呈现下降趋势,说明GlcN的合成速度在前10 h内最快。
在发酵过程中,E. coli-glms-gna1-∆nagE和对照菌株发酵液中的GlcNAc产量均呈现先上升后下降趋势,但对照菌株在发酵后期GlcNAc水平下降较为明显,而E. coli-glms-gna1-∆nagE在后期发酵中GlcNAc水平基本保持在60 g/L以上。E. coli-glms-gna1-∆nagE在培养至 12 h,GlcNAc产量达到最大值71.80 g/L,较对照菌株的GlcNAc产量最高值 (41.50 g/L) 提高了73%。从表3还可看出,对照菌株在残糖低于30 g/L时,GlcNAc产量从最高值41.50 g/L下降到27.00 g/L,下降达35%,而E. coli-glms-gna1-∆nagE在残糖低于10 g/L时,GlcNAc产量从最高值71.80 g/L下降到63.60 g/L,仅下降了11%,这说明nagE基因的敲除在发酵后期能减少GlcNAc向胞内的转运,使发酵液中GlcNAc产量维持稳定。nagE基因的敲除对于提高氨糖的产量效果明显,也说明对照菌株的GlcNAc产量受残糖浓度变化影响显著,而E. coli-glms-gna1- ∆nagE的GlcNAc产量受残糖浓度变化影响不明显。E. coli-glmsgna1-∆nagE和对照菌株的GlcNAc生产强度分别在10 h和6 h时达到最大值,为6.17 g/(L·h) 和6.65 g/(L·h),说明对照菌株在残糖浓度较高时生产强度较高,而E. coli-glms-gna1-∆nagE的生产强度受残糖浓度影响不大。E. coli-glmsgna1-∆nagE单位菌体干重的 GlcNAc产量在10 h达到最高值15.00 g/g DCW,而对照菌株单位菌体干重的GlcNAc产量在6 h达到最高值16.80 g/g DCW,可见对照菌株和E. coli-glmsgna1-∆nagE单位菌体干重的GlcNAc产量相差不明显,E. coli-glms-gna1-∆nagE在残糖浓度为41.00 g/L时的单位菌体干重的GlcNAc产量和对照菌株在残糖浓度为92.00 g/L时的单位菌体干重的GlcNAc产量相近 (分别为15.00 g/g DCW和16.80 g/g DCW),说明残糖浓度的下降对E. coli-glms- gna1-∆nagE单位菌体干重的GlcNAc产量影响不大。
2.3.2 E. coli-glms-gna1-∆nagE-∆manX 发酵结果
从图7和表3可以看出,E. coli-glms-gna1-∆nagE-∆manX的菌浓在 18 h时达到最高值7.03 g/L,而对照菌株的菌浓在14 h时就达到最高值4.68 g/L,这可能与菌株2个基因被敲除,用于合成细胞膜的相关物质的代谢受到影响有关。比较E. coli-glms-gna1-∆nagE和E. coli-glmsgna1- ∆nagE-∆manX的菌浓发现,前者最高值略低于后者,但前者的比生长速率却高于后者,这可能是由于2个基因的敲除在更大程度上抑制了氨基葡萄糖向胞内的转运,细胞比生长速率受到影响,但由于碳源葡萄糖的存在,其转运系统并未受到影响,细胞依然能通过摄取葡萄糖合成自身生长需要的物质。
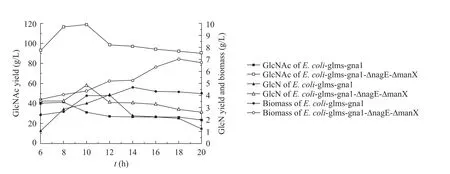
图7 E. coli-glms-gna1-∆nagE-∆manX发酵实验结果Fig. 7 Results of GlcN and GlcNAc production by E. coli-glms-gna1-∆nagE-∆manX.
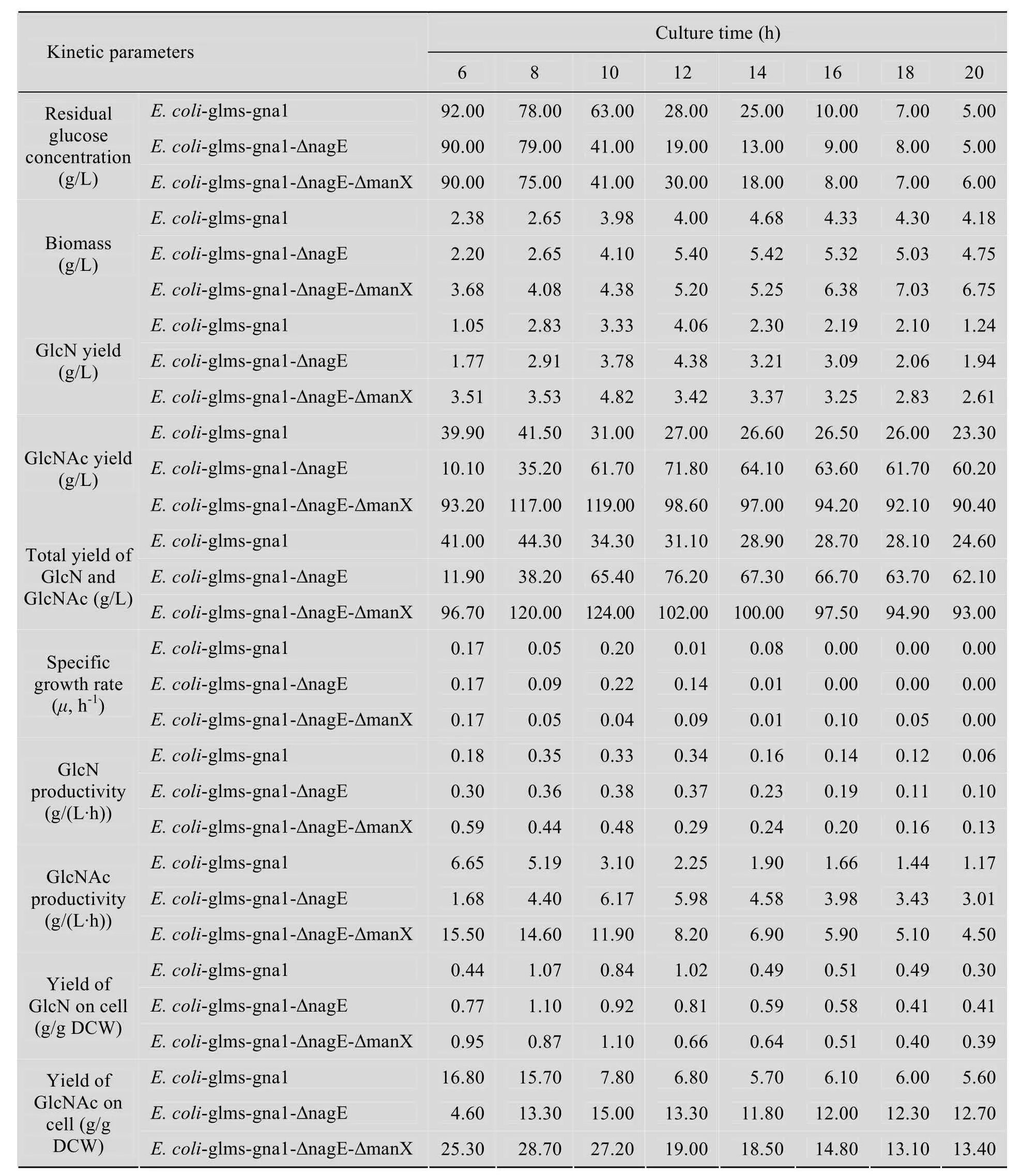
表3 E. coli-glms-gna1-∆nagE, E. coli-glms-gna1-∆nagE-∆manX和E. coli-glms-gna1氨基葡萄糖发酵参数比较Table 3 Comparison of parameters of the glucosamine fermentation by E. coli-glms-gna1-∆nagE, E. coli-glms-gna1- ∆nagE-∆manX and E. coli-glms-gna1
在发酵过程中,E. coli-glms-gna1-∆nagE-∆manX在10 h时GlcN产量达到最大值4.82 g/L,随后呈现缓慢下降趋势,这可能是因为 nagE和manX基因的敲除未能完全抑制GlcN向胞内的转运,细胞生长对碳源的需求导致GlcN能依靠ptsG编码的酶ⅡCBGlc进入细胞,而对照菌株在培养至12 h时GlcN产量达到最高值4.06 g/L。这一方面是由于此时残糖浓度未下降到影响细胞合成GlcN的水平,另一方面是因为此时E. coli-glmsgna1-∆nagE-∆manX的菌浓高于对照菌株的菌浓,通过比较此时单位菌体 GlcN的产量可以发现E. coli-glms-gna1-∆nagE-∆manX单位菌体 GlcN的产量 1.10 g/g DCW 高于对照菌株单位菌体GlcN的产量1.02 g/g DCW。对照菌株的GlcN生产强度在 8 h时达到最高值 0.35 g/(L·h),而E. coli-glms-gna1-∆nagE-∆manX的GlcN生产强度在6 h时达到0.59 g/(L·h),随后呈现下降的趋势。就GlcN产量而言,E. coli-glms-gna1-∆nagE产量的最高值较E. coli-glms-gna1-∆nagE-∆manX低 0.44 g/L,且两者均高于对照菌株,这说明敲除nagE基因能增加葡萄糖向GlcN的代谢,弥补因 GlcNAc不能向胞内转运对细胞生长的影响。
在发酵过程中,E. coli-glms-gna1-∆nagE-∆manX和对照菌株发酵液中的GlcNAc产量均呈现先上升后下降趋势,但 E. coli-glms-gna1-∆nagE-∆manX在发酵后期的GlcNAc水平基本保持在90 g/L以上,在培养至10 h,GlcNAc产量达到最大值 118.70 g/L,较对照菌株的 GlcNAc产量最高值 (41.50 g/L) 提高了 186%,说明manX基因的敲除效果明显,使发酵液中GlcNAc产量维持稳定。E. coli-glms-gna1-∆nagE-∆manX和对照菌株的GlcNAc生产强度均在培养至6 h时最大,分别为15.50 g/(L·h) 和6.65 g/(L·h),随后均呈现下降的趋势。E. coli-glms-gna1-∆nagE-∆manX单位菌体干重的GlcNAc产量在8 h达到最高值 28.70 g/g DCW,而 E. coli-glms-gna1-∆nagE和对照菌株单位菌体干重的 GlcNAc产量分别在10 h和6 h达到最高值15.00 g/g DCW和16.80 g/g DCW,这说明nagE和manX双基因的敲除效果显著。
3 讨论
Red重组敲除技术是近年来兴起的基于λ噬菌体 Red重组酶和体内同源重组反应的新型遗传工程技术,该技术主要利用λ噬菌体的3个重组蛋白酶来完成体外DNA片段与染色体的同源重组。和其他基因敲除技术相比,该技术省去了体外DNA酶切与连接的步骤,使细菌染色体靶基因的敲除与替换操作相对简单,因而逐渐成为基因功能探索以及新工程菌株构建的有效手段。通常对基因的敲除都会对菌体的生长和代谢造成一定的影响,甚至引起菌体死亡。通过比较原菌株与基因敲除菌株的比生长速率以及菌浓发现,基因敲除对菌体生长未出现较大的影响,这可能一方面是与菌体细胞膜合成途径相关联的glmS及gna1基因得到了高效表达,因而对菌体生长起到一定的促进作用;另一方面,胞外的GlcN能通过ptsG编码的酶ⅡCBGlc进入细胞,在碳源不足时起到补偿作用。由于GlcNAc对细胞的刺激远小于 GlcN,这是造成细胞内不能积累GlcN的主要原因之一,所以nagE基因是本研究第一个被敲除的基因。通过对nagE和manX基因的敲除,能提高氨糖的生产强度,或者能缩短菌体达到最大生产强度的时间,这在工业生产上具有较大优势,能充分利用发酵液中的碳源,提高碳源转化率,降低成本,也能简化下游提取工艺,降低污染。对大肠杆菌nagE和manX双基因的敲除,使其 GlcNAc产量达到最大值118.70 g/L,远高于单基因敲除菌株和对照菌株;且双基因的敲除菌株单位菌体干重的GlcNAc产量较对照菌株提高71% (16.80 g/g DCW),较单基因敲除菌株提高91% (15.00 g/g DCW),这说明nagE和manX双基因的敲除能显著提高氨糖的产量,对于改造大肠杆菌发酵生产氨糖具有重要的意义,为最终实现氨糖的工业化生产奠定了坚实的基础。
REFERENCES
[1] Deng MD, Wassink SL, Grund AD. Engineering a new pathway for N-acetylglucosamine production: coupling a catabolic enzyme, glucosamine-6-phosphate deaminase, with a biosynthetic enzyme, glucosamine-6-phosphate N-acetyltransferase. Enzyme Microb Technol, 2006, 39(4): 828−834.
[2] Hsieh JW, Wu HS, Wei YH, et al. Determination and kinetics of producing glucosamine using fungi. Biotechnol Prog, 2007, 23(5): 1009−1016.
[3] Sitanggang AB, Wua HS, Wang SS, et al. Effect of pellet size and stimulating factor on the glucosamine production using Aspergillus sp. BCRC 31742. Bioresour Technol, 2010, 101(10): 3595−3601.
[4] Deng MD, Severson DK, Grund AD, et al. Metabolic engineering of Escherichia coli for industrial production of glucosamine and N-acetylglucosamine. Metab Eng, 2005, 7(3): 201−214.
[5] Sachadyn P, Jędrzejczak R, Milewski S, et al. Purification to homogeneity of Candida albicans glucosamine-6-phosphate synthase overexpressed in Escherichia coli. Protein Expres Purif, 2000, 19(3): 343−349.
[6] Deng MD, Grund AD, Wassink SL, et al. Directed evolution and characterization of Escherichia coli glucosamine synthase. Biochimie, 2006, 88(5): 419−429.
[7] Teplyakov A, Leriche C, Obmolova G, et al. From lobry de bruyn to enzyme-catalyzed ammonia channelling: molecular studies of D-glucosamine-6P synthase. Nat Prod Rep, 2002, 19(1): 60−69.
[8] Sashiwa H, Fujishima S, Yamano N, et al. Production of N-acetyl-D-glucosamine from α-chitin by crude enzymes from Aeromonas hydrophila H-2330. Carbohydr Res, 2002, 337(8): 761−763.
[9] Kuk JH, Jung WJ, Jo GH, et al. Production of N-acetyl-β-D-glucosamine from chitin by Aeromonas sp. GJ-18 crude enzyme. Appl Microbiol Biotechnol, 2005, 68(3): 384−389.
[10] Donzelli BGG, Ostroff G, Harman GE. Enhanced enzymatic hydrolysis of langostino shell chitin with mixtures of enzymes from bacterial and fungal sources. Carbohydr Res, 2003, 338(18): 1823−1833.
[11] Kudan S, Eksittikul T, Pichyangkura R, et al. Preparation of N-acetyl-D-glucosamine and N, N’-diacetylchitobiose by enzymatic hydrolysis of chitin with crude chitinases. J Biotechnol, 2010, 150: 89.
[12] Matsumoto Y, Saucedo-Castañeda G, Revah S, et al. Production of β-N-acetylhexosaminidase of Verticillium lecanii by solid state and submerged fermentations utilizing shrimp waste silage as substrate and inducer. Process Biochem, 2004, 39(6): 665−671.
[13] Chmielowski RA, Wu HS, Wang SS. Scale-up of upstream and downstream operations for the production of glucosamine using microbial fermentation. Biotechnol J, 2007, 2(8): 996−1006.
[14] Rattanakit N, Yano S, Wakayama M, et al. Saccharification of chitin using solid-state culture of Aspergillus sp. Sl-13 with shellfish waste as a substrate. J Biosci Bioeng, 2003, 95(4): 391−396.
[15] Durand P, Golinelli-Pimpaneau B, Mouilleron S, et al. Highlights of glucosamine-6P synthase catalysis. Arch Biochem Biophys, 2008, 474(2): 302−317.
[16] Joanna R, Olchowy J, Konariev PV, et al. The crystal and solution studies of glucosamine-6-phosphate synthase from Candida albicans. J Mol Biol, 2007, 372(3): 672−688.
[17] Beatriz RN, Angel R, Honorina MB, et al. Transport of N-acetyl-D-mannosamine and N-acetyl-D-glucosamine in Escherichia coli K1: effect on capsular polysialic acid production. FEBS Lett, 2002, 511(1/3): 97−101.
[18] Van Montfort RL, Pijning T, Kalk KH, et al. The structure of the Escherichia coli phosphotransferase IIAmannitolreveals a novel fold with two conformations of the active site. Structure, 1998, 6(3): 377−388.
[19] Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA, 2000, 97(12): 6640−6645.
[20] Doublet B, Douard G, Targant H, et al. Antibiotic marker modifications of λ Red and FLP helper plasmids, pKD46 and pCP20, for inactivation of chromosomal genes using PCR products in multidrug-resistant strains. J Microbiol Methods, 2008, 75(2): 359−361.
[21] Su C, Xia WS, Yao HY. The determination of glucosamine and N-acetylglucosamine. Sci Tech Food Ind, 2003, 24(6): 74−75, 77.苏畅, 夏文水, 姚惠源. 氨基葡萄糖和乙酰氨基葡萄糖的测定方法. 食品工业科技, 2003, 24(6): 74−75, 77.
[22] Wang YF, Yan LZ, Guo XQ. Determination of N-acetyl-D-glucosamine in D-glucosamine hydrochloride by HPLC with refractive index detector. J Cap Norm Univ: Nat Sci Ed, 2008, 29(1): 40−42.王英锋, 晏利芝, 郭雪清. HPLC示差折光分析法测定D-氨基葡萄糖盐酸盐中的N-乙酰-D-氨基葡萄糖. 首都师范大学学报: 自然科学版, 2008, 29(1): 40−42.
猜你喜欢
睿士(2023年9期)2023-09-20 05:47:07
当代水产(2022年1期)2022-04-26 14:35:38
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
现代畜牧科技(2021年9期)2021-10-13 06:38:40
生物工程学报(2019年1期)2019-01-30 08:19:58
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
小雪花·初中高分作文(2016年9期)2016-05-14 02:50:08
精细石油化工(2015年3期)2015-12-14 09:07:42
江西理工大学学报(2013年1期)2013-03-20 14:57:07
