
组蛋白去乙酰化酶1、3在炎症牙周膜中的表达
2012-02-05 01:48刘大勇赵梦明荆晓艳
牙体牙髓牙周病学杂志 2012年11期
肖 蕊,刘大勇,赵梦明,荆晓艳,贾 智
(天津医科大学口腔医院,天津300070)
组蛋白乙酰化属于表观遗传学的范畴,是一种不涉及DNA序列改变的可遗传的基因表达变化过程[1]。组蛋白乙酰化状态受两类酶 — 组蛋白乙酰化转移酶(histone acetyltransferases,HAT)和组蛋白去乙酰化酶(histone deacetylases,HDAC)的调节[2]。组蛋白乙酰化和去乙酰化作用是调节炎症反应中基因表达的重要过程,有研究表明,HDAC是参与炎症基因转导和细胞增殖的关键因子[3]。哺乳类HDAC可分为4组,共18种:组I (HDAC 1、HDAC 2、HDAC 3、HDAC 8)在大多数细胞中普遍存在,主要位于细胞核,调控组蛋白的乙酰化修饰;组Ⅱ(HDAC 4、HDAC 5、HDAC 6、HDAC 7、HDAC9、HDAC10)常频繁穿梭于细胞核内和胞浆之间,调控组蛋白和非组蛋白的乙酰化修饰;组Ⅲ(Sirt 1、Sirt 2、Sirt 3、Sirt 4、Sirt 5、Sirt 6、Sirt 7)能够使p53去乙酰化,从而导致p53介导的转录、凋亡等一系列生物学活动失去作用;组Ⅳ(HDACll)可能与免疫耐受有关。
牙周炎是牙周组织慢性感染性疾病,是成人失牙的主要原因,并且与许多全身性病相关,如糖尿病、心血管疾病、新生儿早产和低体质量儿等。牙周炎的发生过程是以牙周致病菌为始动因子而引起的机体过度免疫反应,并最终导致牙周组织破坏。研究证实,牙周炎免疫反应具有自身免疫反应的特征[4],且在发病机制、治疗等方面与类风湿性关节炎有许多相似之处[5-6]。
与骨关节炎和正常滑膜组织相比,类风湿性关节炎中的HDAC 1过表达[7]。研究表明[8]:一种新型HDAC 3选择性抑制剂-MI192能减少类风湿性关节炎患者外周血单核细胞(PBMC)中IL-6的产生,从而缓解其免疫炎症反应。然而,牙周炎的发生与牙周组织中乙酰化状态的改变是否有关尚未见报道。本研究通过比较炎症牙周膜与健康牙周膜中HDAC 1、HDAC 3的表达,初步探讨HDAC与牙周炎发病的关系。
1 材料和方法
1.1 主要试剂和仪器
TransZol Up、TransScript Two-Step RT-PCRsuperMix、DNAmarker(全式金,北京);引物(上海生工);琼脂糖、梯度PCR仪(MJ Research公司,美国);电泳凝胶成像仪(Vitbertourmat公司,法国)。
1.2 组织样本采集
所选病例均来自天津医科大学口腔医院门诊患者,实验组选择20~55岁慢性牙周炎患者20例,纳入标准:①探诊深度(PD)≥5 mm,临床附着水平(CAL)≥5 mm,X线片示牙槽骨吸收≥根长的1/2;②牙齿松动≥Ⅲ度,需要拔除患牙。排除标准:①药物、妊娠等导致的牙龈增生;②患有糖尿病等全身性病;③吸烟者。对照组选择20~55岁牙周健康[探诊深度(PD)≤3 mm,无附着丧失,无牙龈出血或红肿]者20例,均无全身性病,不吸烟且有需要拔除的正畸减数牙或智齿。收集符合以上标准的所有受试者被拔除的牙齿,并刮取根中1/3牙周膜组织置无酶无菌的冻存管内,液氮罐中保存备用。
1.3 RT-PCR检测炎症及健康牙周膜中HDAC 1,HDAC 3的表达
1.3.1 引物合成
HDAC 1上游引物:5'-AGCCAAGAGAGTCAAAACAGA-3';下游引物:5'-GGTCCATTCAGGCCAACT-3';产物长度102 bp。HDAC 3上游引物:5'-TGGCTTCTGCTATGTCAACG-3';下游引物:5'-GCACGTGGGTTGGTAGAAGT-3';产物长度308 bp。GAPDH上游引物:5'-GTCAGTGGTGGACCTGACCT-3';下游引物:5'-AGGGGAGATTCAGTGTGGTG-3';产物长度413 bp。以上引物均由上海生工生物技术公司合成。
1.3.2 提取总RNA
将上述冻存的组织样本转移至液氨预冷的研钵中,研磨至粉状后,每50~100 mg组织中加入1 mL TransZol继续研磨。然后转移至EP管,室温静置 5 min,加入 0.2 mL氯仿剧烈震荡,12 000 r/min 4℃离心15 min,取无色水相上层;加入0.5 mL异丙醇析出沉淀,12 000 r/min 4℃离心10 min,弃上清;用1 mL无RNA酶的750 mL/L乙醇洗涤沉淀,7 500 r/min 4℃离心5 min,弃上清,室温干燥形成胶状沉淀。将胶状沉淀用10 μL RNA溶解液溶解后,取1 μL稀释100倍,用紫外分光光度计分别测定280 nm和260 nm处的吸光度值(OD),以评定其浓度和纯度。最后根据测定值调整RNA标本浓度为1 μg/μL。
1.3.3 cDNA第一链合成和多聚酶链反应(PCR)
严格按照TransScript Two-Step RT-PCR super-Mix试剂盒说明进行。首先合成cDNA第一链,合成体系20 μL:总RNA50 ng~5 μg;Anchored OLigo 1 μL;2×TS Reaction Mix 10 μL;TransScript RT/RI Enzyme Mix 1 μL,Rnase-free Water to 20 μL。然后PCR扩增HDAC 1、HDAC 3和内参GAPDH。
PCR反应体系25 μL:cDNA 1 μL;上游引物1 μL;下游引物1 μL;2×TransTaqTM HiFi PCR SuperMix 12.5 μL;ddH2O 8.5 μL。反应条件为: 94℃变性5 min后,94℃变性30 s,HDAC1,56℃; HDAC 3,59℃;GAPDH,58℃退火30 s,72℃延伸1 min,35个循环后72℃再延伸10 min。PCR产物行溴化乙啶染色(EB染色)琼脂糖凝胶电泳,观察并照相。
1.3.4 灰度值分析
用Quantity One软件对电泳图进行灰度值分析,以样品灰度值与同一样品GAPDH的灰度值之比作为评价HDAC 1,HDAC 3表达量的指标。
1.4 统计学分析
用SPSS 16.0统计软件进行统计分析,实验组和对照组比较用独立样本的t检验,检验水准α= 0.05。
2 结果
在炎症牙周膜和健康牙周膜组织中均有HDAC1、HDAC 3表达(图1)。经灰度值分析,HDAC1、HDAC3在炎症牙周膜中的表达均明显高于健康牙周膜,差异有统计学意义(P<0.05)(图2)。
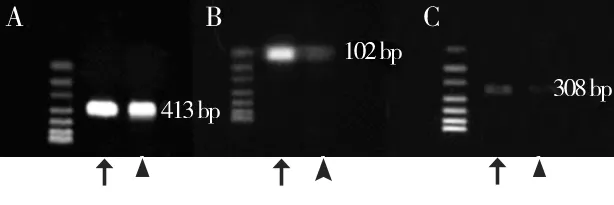
图1 炎症和健康牙周膜中HDAC 1、HDAC 3的表达
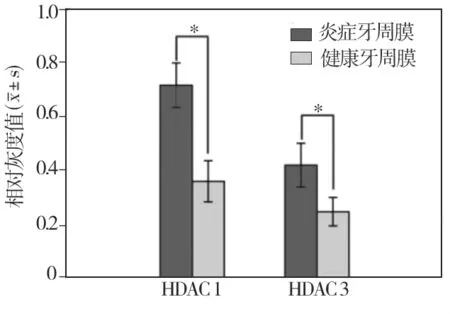
图2 炎症、健康牙周膜中HDAC 1、HDAC 3相对灰度值比较
3 讨论
由于核心组蛋白富含带正电荷的碱性氨基酸,与DNA具有高度亲和性,能阻碍基本转录单位蛋白质复合物进入启动子结合位点,而导致转录功能受到抑制。组蛋白氨基末端特定部位ε-氨基酸的乙酰化可中和其正电荷,减弱核小体中碱性氨基酸与DNA的静电吸引力,增加转录因子的进入,从而促进基因的转录。因此,组蛋白的乙酰化和去乙酰化与基因的表达调控密切相关。负责组蛋白乙酰化和去乙酰化的是两种功能相互拮抗的蛋白酶HAT和HADC,两者之间的动态平衡控制着染色质的结构和基因的表达。当HDAC过度表达并被转录因子募集,就会导致特定基因的不正常抑制,从而导致炎症性自身免疫病等其他疾病。
乙酰化修饰在炎症性自身免疫性疾病中起重要作用,其中包括组蛋白乙酰化修饰和非组蛋白乙酰化修饰。组蛋白乙酰化修饰是基因转录激活的重要标志。当炎症信号传至核内、转录因子与启动子结合后,可进一步募集P300/CBP等共刺激因子,他们作用于组蛋白N端的赖氨酸残基使其发生乙酰化修饰[9]。非组蛋白的乙酰化修饰包括NF-КB等的乙酰化修饰。近期研究发现,乙酰化等翻译后修饰在NF-КB的亚细胞定位、DNA结合力、转录活性等方面起重要调节作用[10]。当炎症刺激因子作用于机体时,P38等MAPK信号通路的激活可磷酸化P300并激活HAT活性,还可乙酰化P65,增加NF-КB与КB序列的结合能力启动NFКB介导的炎症相关基因转录[11]。IL-1、IL-6启动子上均有NF-КB结合位点,其基因转录受NF-КB调控,NF-КB活化后促进炎症因子基因转录从而增加炎症相关蛋白的表达[12]。有研究发现: HDAC 1、HDAC 3可直接或间接与NF-КB结合,在NF-КB去乙酰化中发挥一定作用[13-14]。
本结果显示:牙周炎患者牙周膜中HDAC 1、HDAC 3 mRNA表达明显高于牙周健康者(与类风湿性关节炎研究类似[7-8]),打破了组蛋白乙酰化和去乙酰化的动态平衡,有可能促进了NF-КB介导的炎症因子的表达,参与了牙周炎症反应的基因调控。
研究证实:由于可以减少炎症因子的产生,组蛋白去乙酰化酶抑制剂(histone deactylase inhibitor,HDACi)在体内外具有免疫调节作用[15-16]。HDACi可以调节NF-КB信号通路,从而降低炎症因子和转录因子的水平[17-18]。HDACi已被用于治疗多种炎症性自身免疫病,包括类风湿性关节炎、哮喘、银屑病、多发性硬化病、系统性红斑狼疮等[19-20]。局部用药能降低系统毒性和有关的副作用[21]。局部应用组蛋白去乙酰化抑制剂可能会为牙周炎治疗开辟一条新途径。
总之,本研究发现HDAC 1、HDAC 3在炎症牙周组织中高表达,该结果虽初步从表观遗传学的角度探索了牙周炎可能的发病机制,但组蛋白乙酰化和去乙酰化过程调控牙周炎症免疫反应的确切机制,以及组蛋白去乙酰化酶抑制剂能否成为牙周炎治疗的潜在药物,还需进一步研究证实。
[1] Goldberg AD,Allis CD,Bernstein E.Epigenetics:a landscape takes shape[J].Cell,2007,128(4):635-638.
[2] Khochbin S,Verdel A,Lemercier C,et al.Functional significance of histone deacetylase diversity[J].Curr Opin Genet Dev,2001,11(2):162-166.
[3] Adcock IM,Ford P,Ito K,et al.Epigenetics and airways disease[J].Respir Res,2006,7:21.
[4] Nesse W,Dijkstra PU,Abbas F,et al.Increased prevalence of cardiovascular and autoimmune diseases in periodontitis patients:a cross-sectional study[J].J Periodontol,2010,81 (11):1622-1628.
[5] Sweier DG,Shelburne PS,Giannobile WV,et al.Immunoglobulin G(IgG)class,but Not IgA or IgM,antibodies to peptides of the Porphyromonas gingivalis chaperone HtpG predict health in subjects with periodontitis by a fluorescence enzyme-linked immunosorbent assay[J].Clin Vaccine Immunol,2009,16 (12):1766-1773.
[6] Liao F,Li Z,Wang Y,et al.Porphyromonas gingivalis may play an important role in the pathogenesis of periodontitis-associated rheumatoid arthritis[J].Med Hypotheses,2009,72(6): 732-735.
[7] Kawabata T,Nishida K,Takasugi K,et al.Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis[J].Arthritis Res Ther,2010,12(4):R133.
[8] Gillespie J,Savic S,Wong C,et al.Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients[J].Arthritis Rheum,2012,64(2):418-422.
[9] Berndsen CE,Denu JM.Catalysis and substrate selection by histone/protein lysine acetyltransferases[J].Curr Opin Struct Biol,2008,18(6):682-689.
[10] Pan WW,Li JD,Huang S,et al.Synergistic activation of NF-{kappa}B by bacterial chemoattractant and TNF{alpha}is mediated by p38 MAPK-dependent RelA acetylation[J].J Biol Chem,2010,285(45):34348-34354.
[11] Saha RN,Jana M,Pahan K.MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65[J].J Immunol,2007,179(10):7101-7109.
[12] Blackwell TS,Christman JW.The role of nuclear factor-kappa B in cytokine gene regulation[J].Am J Respir Cell Mol Biol,1997,17(1):3-9.
[13] Spange S,Wagner T,Heinzel T,et al.Acetylation of non-histone proteins modulates cellular signalling at multiple levels[J].Int J Biochem Cell Biol,2009,41(1):185-198.
[14] Ashburner BP,Westerheide SD,Baldwin AJ.The p65(RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC)corepressors HDAC1 and HDAC2 to negatively regulate gene expression[J].Mol Cell Biol,2001,21(20):7065-7077.
[15] Leoni F,Zaliani A,Bertolini G,et al.The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines[J].Proc Natl Acad Sci U S A,2002,99(5):2995-3000.
[16] Leoni F,Fossati G,Lewis EC,et al.The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo[J].Mol Med,2005,11(1-12):1-15.
[17] Ghosh S,Hayden MS.New regulators of NF-kappaB in inflammation[J].Nat Rev Immunol,2008,8(11):837-848.
[18] Gupta SC,Sundaram C,Reuter S,et al.Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy[J].Biochim Biophys Acta,2010,1799(10-12):775-787.
[19] Velazquez OC,Lederer HM,Rombeau JL.Butyrate and the colonocyte.production,absorption,metabolism,and therapeutic implications[J].Adv Exp Med Biol,1997,427:123-134.
[20] Vernia P,Annese V,Bresci G,et al.Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis:results of a multicentre trial[J].Eur J Clin Invest,2003,33(3):244-248.
[21] Choo QY,Ho PC,Lin HS.Histone deacetylase inhibitors:new hope for rheumatoid arthritis?[J].Curr Pharm Des,2008,14 (8):803-820.
猜你喜欢
中国运动医学杂志(2022年8期)2022-11-25
中国生物化学与分子生物学报(2022年8期)2022-09-08
中国药学药品知识仓库(2022年10期)2022-05-29
中国药学药品知识仓库(2022年10期)2022-05-29
健康体检与管理(2022年4期)2022-05-13
中国典型病例大全(2022年10期)2022-05-10
复旦学报(自然科学版)(2021年5期)2021-11-17
昆明医科大学学报(2021年4期)2021-07-23
现代临床医学(2021年2期)2021-03-29
福建轻纺(2020年3期)2020-11-29
