
Purification and Characterization of an Extracellular Manganese Peroxidase from Trametes gallica
2012-01-12 06:58SUNXunZHUTaoZHANGYizheng
微生物学杂志 2012年5期
SUN Xun,ZHU Tao,ZHANG Yi-zheng
(1.Life Science Department,Heze University,Heze 274015,China;2.Life Science College,Sichuan University,Chengdu 610064,China)
It is well known that wood is mainly composed of cellulose,hemicellulose and lignin,and that it is the most abundant and renewable bio-resource in nature.White rot fungi are the most efficient ligninolytic microorganisms in nature,and most of them,for instance Phanerochaete chrysosporium(Tien,1987)and Trametes versicolor(Paice et al.,1993),secrete several lignin degrading enzymes including lignin peroxidase(LiP),laccase(Lac),and manganese dependent peroxidase(MnP).MnP is the most common ligninolytic peroxidase produced by almost all white-rot basidiomycetes.This enzyme catalyzes the oxidation of Mn(Ⅱ)to Mn(Ⅲ),which in turn can oxidize phenolic substrates(Wariishi et al.,1988).The remarkable degradative potential of MnP makes this enzyme an attractive and versatilebiocatalyst for biotechnological and environmental applications,e.g.in the pulping and bleaching of lignocellulose and in the removing of hazardous wastes(Wesenberg et al.,2003;Hofrichter et al.,2010).A lot is already known from manganese peroxidases in white-rot basidiomycetes,and many genes have been described and characterized,however,in order to facilitate the potential use of these enzymes in biotechnological applications,there is space to explore these enzymes in variable sources and characterize novel variants.For such goals,basic characterizations of MnPs are well worth exploring.Our previous study showed that the white-rot basidiomycete Trametes gallica secreted all the ligninolytic peroxidases referenced above and had a strong capacity to degrade lignocelluloses(Sun et al.,2002;Sun et al.,2004).In this study,we firstly isolated and purified an extracellular MnP from the fungus cultured on wheat straw powder.
1 Materials and Methods
1.1 Organism and culture conditions
The strain used in this study was initially isolated in 1997,from a poplar stump in Heze,China,preliminarily identified as Trametes gallica Fr.,and collected at Molecular Biology Lab of Heze University.The strain was maintained at 4℃on potato dextrose agar(PDA)slants.One liter of nutrient salt fluid,devoid of a carbon source,contained the following ingredients:3.0gammoniumtartrate,KH2PO41.0 g,MgSO4·7H2O 0.5 g,CaCl2·2H2O 0.1 g and trace mineral solution 2 mL(MnSO4·H2O 0.5 g,FeSO4·7H2O 0.5 g,CuSO4·5H2O 0.6 g,ZnSO4·7H2O 0.05 g,H3BO30.2 g,Na2MoO4·2H2O 0.2 g,CoCl2·6H2O 0.1 g,distilled H2O 1 000 mL).Air-dried wheat straw powder(250 g,in 60-mesh)was mixed with 750 mL of the nutrient salts fluid,equally distributed in 10 500 mL Erlenmeyer flasks and autoclaved for 1.5 h at 121℃.Three mycelial disks(1 cm in diam.)were punched from the fungus grown on PDA plate for 6 days at 26℃and used as inocula for each culture.Each flask was sealed with eight-layer sterile gauze for air exchange and cultured for 30 days at 26℃.
1.2 Preparation of crude enzyme
The harvested cultures were gently scattered,mixed with 3 000 mL sterile dH2O,extracted for 4 h at 4℃in a shaker(120 r/min)and filtered through 3 layers of gauze.The residue was discarded and the filtrate was further centrifuged(8 000 g,10 min)to remove any remaining solids.(NH4)2SO4powder was added to the filtrate(2 500 mL)to 30%saturation to remove protein precipitate through centrifugation(8 000 g,15 min).The(NH4)2SO4was then increased to 80%saturation in the supernatant and incubated overnight at 4℃,after which the solution was centrifuged(10 000 g,20 min,4℃)again.The precipitate was dissolved in dH2O and dialyzed(molecular weight cutoff value of 12~14 ku)against dH2O.The resulting crude enzyme was subjected to further purification.
1.3 Protein and enzyme assays
Protein content was estimated by Coomassie-dye binding method(Spector,1978).LiP(EC 1.11.1.14)activity was measured as described by Cai and Tien(1991)by monitoring the increase at A310 associated with the oxidation of veratryl alcohol to veratraldehyde.One unit(U)of LiP was defined as the amount of enzyme that formed 1 μmol veratraldehyde per minute(ε=9 300 M-1cm-1).Manganese(Ⅱ)peroxidase(MnP)(EC 1.11.1.13)activity was assayed according to the slightly modified method of Wariishi(1988).A suitable volume of MnP-containing liquid was added to the reaction mixture consisting of 100 mmol/L sodium lactate(pH 5.0),0.1 mmol/L MnSO4and 0.1 mmol/L H2O2.Reactions were initiated by addition of H2O2.Oxidation of Mn(Ⅱ)to Mn(Ⅲ)was measured at 30℃by the increase in A240 where the complex formed between Mn3+and lactate was monitored(ε=6 500 M-1cm-1).One unit(U)of MnP activity was defined as the amount of enzyme required to oxidize 1 μmol Mn2+to Mn3+per minute.Laccase(EC 1.10.3.2)activity was determined by using ABTS 2,2'-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)as substrate.Oxidation of ABTS was monitored at 30℃by measuring the increase in A420(ε=36 000 M-1cm-1).One unit(U)of laccase activity was defined as the amount of enzyme required to oxidize 1 μmol of ABTS per minute(Wolfenden and Willson,1982).β-Xylanase assay was carried out by dinitrosalicylic acid(DNS)reagent(Miller,1959),using D-xylose as a standard.The reaction mixture,which contained 0.1 mL of enzyme sample and 0.9 mL of 1%birch wood xylan(Sigma Chem.Co.)in 50 mmol/L sodium acetate buffer(pH 4.5),was incubated at 45℃for 30 min.One unit of β-xylanase was defined as the amount of enzyme that liberated from xylan 1 μmol equivalent of xylose in one minute.The assay for β-(1→4)D-glucanase activity was equal to that for xylanase except that the substrate was CMC(Sigma),and D-glucose was used as a standard instead of xylose.
1.4 Ion-exchange chromatography and gel filtration
Ion exchange was performed using a Q-Sepharose FF packed column(20 cm×1.6 cm,Pharmacia).The column was equilibrated with 10 mmol/L sodium acetate buffer(pH 5.5).The proteins adsorbed to the column were released through elution with a sodium chloride gradient(0 to 1 mol/L)prepared in the same buffer.The enzyme activities in each fraction(3 mL)were tested as described above.The fractions with the same enzyme were collected respectively,added with ammonium sulphate(80%saturation),incubated overnight at 4℃and centrifuged(10 000 g,4℃,20 min).The harvested protein precipitate was dissolved in 20 mmol/L sodium acetate buffer(pH 5.5)and dialyzed overnight against 2 liters of the same buffer.The dialysate was transferred into a dialysis bag and concentrated with polyethylene glycol(PEG 20 000).The concentrated solution was used as the crude enzyme source for the next step of the chromatography.
A column(100 cm×1.6 cm)packed with Sephadex G-100 was employed to further purify MnP.The column was equilibrated with 20 mmol/L sodium acetate buffer(pH 5.5).Fractions of 1 mL(linear flow rate of 6 cm/h)were collected and enzyme activities were detected by the corresponding enzyme assay.The fractions with the same enzyme were respectively merged and further treated as described above.
1.5 Analytical gel electrophoresis and recovery of enzymes
The partially purified enzymes from Sephadex G-100 chromatography were further analyzed and recovered using native polyacrylamide gel electrophoresis(PAGE).The native PAGE was performed by the method of Laemmli(1970),with some modifications(emission of sodium dodecyl sulfate in gel and β-mercaptoethanol in the loading sample).To visualize MnP,the gel was directly immersed in 2 mmol/L guaiacol prepared in 100 mmol/L lactic acid buffer(pH 5.0),which contained 0.1 mmol/L MnSO4·H2O and 0.1 mmol/L H2O2,and incubated at 37℃.
Recovery of MnP in the running gel was achieved as follows.Cut two 1 cm wide slices at both sides of the gel,incubate and visualize using the method mentioned above;reset the two slices and the remaining gel;in accordance with the enzyme spectrum on the two gel slices,horizontally cut the remaining gel,collect the enzyme-containing gel slice,and transfer it into a dialysis bag(12~14 ku molecular weight cutoff);electro-dialyze for 2~3 hours(15 mA,4℃)in a horizontal electrophoresis tank containing 25 mmol/L Tris-glycine buffer,pH 8.3;collect the dialysate and further dialyze against dH2O,thence the purified enzyme can be harvested.Concentration of the enzyme solution using PEG(Mw.20 000)was necessary if the quantitative analysis of the purified enzyme(e.g.determination of molecular weight and isoelectric point)further proceeded.
1.6 Determination of molecular weight and isoelectric point of proteins
SDS-PAGE was carried out in 8%polyacrylamide gel slabs at pH 8.3 using 25 mmol/L Tris-glycine buffer containing 0.1%(w/v)SDS according to Laemmli(1970).The low molecular mass cali-bration mixture was used as a standard.Isoelectric focusing(IEF)was carried out in Mini-ProteanⅡGel System(Bio-Rad)according to the manufacturer’s instructions,using Ampholytes 2.80~6.55(Low pI Kit,Amersham,Pharmacia)and 4.45~9.60(Bio-Rad).
2 Result and discussion
2.1 Enzyme purification
The ion-exchange elution profile of laccase,MnP and LiP is given in Fig.1 .The fractions showing LiP activities were completely included in the MnP fractions,although the LiP peak is slightly ahead of MnP peak(Fig.1 ).The fractions showing laccase activity were distributed from tube No.20 to 70(Fig.1 ),potentially indicating production of a variety of laccase isoenzymes with different isoelectric points.It had been reported that this strain could produce a total of 20 laccase isoenzymes(Dong et al.,2005).As the conventional ion exchange chromatography is of low resolution,it is difficult to clearly distinguish isozymes of an enzyme from its eluting curve.
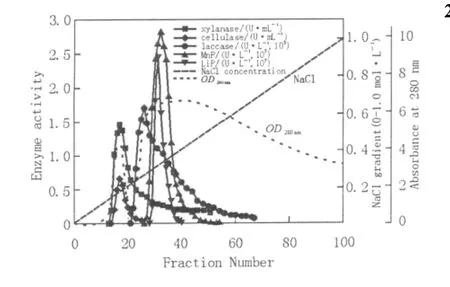
Fig.1 Q-Sepharose chromatography of lignocellulolytic enzymes from Trametes gallica Fr
The fractions exhibiting laccase,MnP and LiP activities from the Q-Sepharose column were pooled,salted-out,concentrated with PEG and further applied to a Sephadex G-100 column.The laccase peak is ahead of MnP and LiP,suggesting that laccase has a greater molecular weight than MnP and LiP(Fig.2 ).LiP and MnP were almost on the same elution peak;however,the LiP peak is slightly ahead of MnP,indicating that LiP has a slightly greater molecular weight than MnP.The results showed that the recovery rate and purification fold of MnP were 28%and 27.3,respectively.The fractions from Sephadex G-100,which exhibited MnP was further purified by using native-PAGE.

Fig.2 Sephadex G-100 column chromatogram of laccase,MnP and LiP
2.2 Properties of the purified MnP
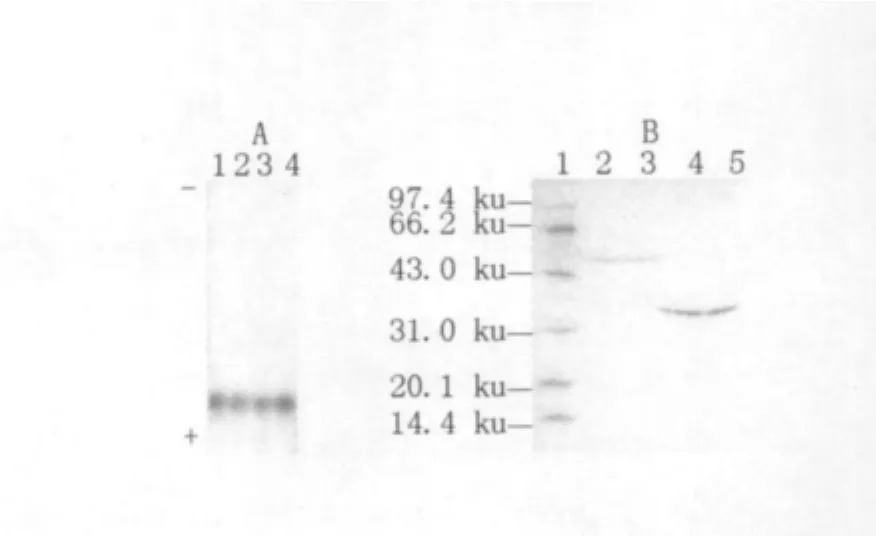
Fig.3 The native-PAGE and SDS-PAGE of the purified MnP
The recovered MnP from the native PAGE showed a single band in native PAGE gel and SDSPAGE gel,respectively,meaning that the enzyme had been purified in the study(Fig.3 ).The molecular weights of MnPs in white rot fungi are generally about 45 ku,but MnP isoenzymes in Pleurotus ostreatus were only 36.4 ku(Ha et al.,2001).The purified MnP,which had a molecular weight of approximately 35.7 ku in the study(Fig.3 ),was possibly a single-chain protein,since MnPs in white rot fungi were mostly single-chain proteins(Gold and Alic,1993).
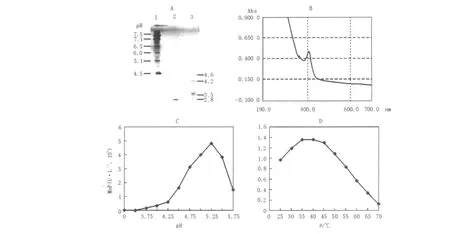
Fig.4 Some properties of the purified MnP
The isoelectric point of the purified MnP was evaluated as pI 2.8(Fig.4 A).Many white rot fungi such as Phanerochaete chrysosporium(Pribnow et al.,1989;Godfrey et al.,1990;Datta et al.,1991),Phanerochaete flavido-alba(Ruiz et al.,2002)and Pleurotus ostreatus(Sarkar et al.,1997;Ha et al.,2001)produced manganese peroxidases with pI 3.5~5.0.The purified MnP was heme glycoprotein exhibiting a maximal absorbance at 407 nm in the study(Fig.4 B).
The effect of pH on MnP activity was measured over the range 3.5~5.75 using a 100 mmol/L sodium lactate buffer system at 37℃,with optimum activity occurring at pH 5.25(Fig.4 C).The temperature optimum was determined within the temperature range 25~70℃in 100 mmol/L sodium lactate buffer at pH 5.25.The temperature-activity profile of the purified MnP showed a curve with one peak at about 35℃(Fig.4 D).
Reference
[1] Cai,D.Y.,M.Tien.Lignin peroxidase of Phanerochaete chrysosporium[J].J.Biol.Chem,1991,266(22):14464-14469.
[2] Datta,A.,A.Bettermann,T.K.Kirk.Identification of a specific manganese peroxidase among ligninolytic enzymes secreted by Phanerochaete chrysosporium during wood decay[J].Appl.Environ.Microbiol,1991,57(5):1453-1460.
[3] Dong,J-L.,Y-W.Zhang,R-H.Zhang,et al.Influence of culture conditions on laccase production and isozyme patterns in the white-rot fungus Trametes gallica[J].J.Basic Microbiol,2005,45(3):190-198.
[4] Godfrey,B.J.,M.B.Mayfield,J.A.Brown,et al.Char-acterization of a gene encoding a manganese peroxidase from Phanerochaete chrysosporium[J].Gene,1990,93(1):119-124.
[5] Gold,M.H.,M.Alic.Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium[J].Microbiol.Rev,1993,57(3):605-622.
[6] Ha,H.C.,Y.Honda,T.Watanabe,et al.Production of manganese peroxidase by pellet culture of the lignin-degrading basidiomycete,Pleurotus ostreatus[J].Appl.Microbiol.Biotechnol,2001,55(6):704-711.
[7] Hofrichter,M.,R.Ullrich,M.J.Pecyna,et al.New and classic families of secreted fungal heme peroxidases[J].Appl.Microbiol.Biotechnol,2010,87(3):871-897.
[8] Laemmli,U.K.Cleavage of structural proteins during the assembly of the head of bacteriophage T4[J].Nature,1970,227:680-685.
[9] Miller,G.L.Use of dinitrosalicylic acid reagent for determination of reducing sugar[J].Anal.Chem,1959,31:426-428.
[10] Paice,M.G.,I.D.Reid,R.Bourbonnais,et al.Jurasek.Manganese peroxidase,produced by Trametes versicolor during pulp bleaching,demethylates and delignifies kraft pulp[J].Appl.Environ.Microbiol,1993,59(1):260-265.
[11] Pribnow,D.,M.B.Mayfield,V.J.Nipper,et al.Characterization of a cDNA encoding a manganese peroxidase,from the lignin-degrading basidiomycete Phanerochaete chrysosporium[J].J.Biol.Chem,1989,264(9):5036-5040.
[12] Ruiz,J.C.,T.de la Rubia,J.Pérez,et al.Effect of olive oil mill wastewater on extracellular ligninolytic enzymes produced by Phanerochaete flavido-alba[J].FEMS Microbiol.Lett,2002,212(1):41-45.
[13] Sarkar,S.,A.T.Martinez,M.J.Martinez.Biochemical and molecular characterization of a manganese peroxidase isoenzyme from Pleurotus ostreatus[J].Biochim.Biophys.Acta,1997,1339(1):23-30.
[14] Spector,T.Refinement of the Coomassle blue method of protein quantitation[J].Anal.Biochem,1978,86(1):142-146.
[15] Sun,X.,S-T.Ren and R-M.Bi.Lignocellulose degradation of wheat straw by Trametes gallica[J].J.Microbiol.(in Chinese),2002,22(1):24-26.
[16] Sun,X.,R.Zhang,Y.Zhang.Production of lignocellulolytic enzymes by Trametes gallica and detection of polysaccharide hydrolase and laccase activities in polyacrylamide gels[J].J.Basic Microbiol,2004,44(3):220-231.
[17] Tien,M.Properties of ligninase from Phanerochaete chrysosporium and their possible applications[J].Crit.Rev.in Microbiol,1987,15(2):141-168.
[18] Wariishi,H.,L.Akileswaran,M.H.Gold.Manganese peroxidase from the basidiomycete Phanerochaete chrysosporium:spectral characterization of the oxidized states and the catalytic cycle[J].Biochem,1988,27(14):5365-5370.
[19] Wesenberg,D.,I.Kyriakides,S.N.Agathos.White-rot fungi and their enzymes for the treatment of industrial dye effluents[J].Biotechnol.Adv,2003,22:161-187.
[20] Wolfenden,B.S.,R.L.Willson.Radical cations as reference chromogens in kinetic studies of one-electron transfer reactions;pulse radiolysis studies of 2,2'-azinobis-(3-ethylbenzothiazoline-6-sulfonate)[J].J.Chem.Soc,1982,24:805-812.
