
含苯巯基分子电子输运性质的研究*
2011-12-17 09:41:50童国平
浙江师范大学学报(自然科学版) 2011年3期
黄 埔, 童国平
(浙江师范大学数理与信息工程学院,浙江金华 321004)
0 引言
近年来,随着纳米科学研究的不断深入,分子器件的发展引人关注,实验工作者利用各种测量技术对分子器件进行研究,发现分子具有许多重要的电学特性[1-3].1997年,Reed研究组[1]第一次成功地测量了单个分子的伏安特性,揭示了分子的输运特征.理论上模拟分子器件工作原理的方法有多种[4-8],使分子结构或电子结构与电学性质联系起来.Mujica等[4]及Tian等[6]改进了研究分子结构电输运性质的方法,他们的理论模型能更好地应用格林函数来解决问题,尤其是以苯环为基本结构单元的芳香族有机化合物,对其导电性质的研究备受大家的关注.在苯环中,碳原子最外层的电子发生s杂化,每个碳原子都可以提供一个自由移动的π电子,对分子的导电起重要的作用,因此,基于苯基结构的芳香族有机化合物成为分子电子学研究中的重点.尽管实验上对含苯环分子有较多的研究[9-16],但理论研究中对含不同数目的苯环巯基分子的系统研究还较少.因此,研究苯环数目,特别是研究在非平衡态下的苯环数目对分子器件电输运特性的影响有着极其重要的作用,它能够为更好地理解和预测分子器件的导电特性打下坚实的基础.本文以实验中所研究的含苯分子为对象,从第一性原理出发,利用密度泛函和非平衡格林函数理论方法,研究了含不同数目苯环的巯基分子的电子结构及电输运性质,同时讨论了苯环数目对分子几何结构的影响,比较了在不同的几何结构下分子的电输运性质.
1 模型和方法
笔者以文献[17]中提出的模型为基础,建立了如图1所示的含苯环数目递增的1组模型来模拟不同苯环数目对分子器件电输运特性的影响.本文选用Au作为金属电极,构造了3个电极-分子-电极的桥式结构形式.考虑到分子与电极相互作用的局域性[18-19]等因素,选用4×4的Au(111)面模拟半无限大电极与分子间的相互作用[17].Au与Au之间的距离设定为Au的晶格常数0.288 nm,自由分子的两端S原子处于3个Au原子组成的正三角形的中心的正上方,也就是 Hollow 位置上[1,18,20],通过 S 原子化学吸附在Au表面,一起组成扩展分子.改变电极距离,系统能量最低时即为电极稳定距离.整个系统包括左电极、中心区域和右电极3个部分,如图1所示.其中中心区域由含苯分子和2层Au原子组成,在中心区域中,与分子连接的 2层Au(111)面与左右电极采用相同的模型,用相同的参数进行描述[20],研究苯环数目对体系电输运性质的影响.应用Landauer-Büttiker公式可求出体系的电流
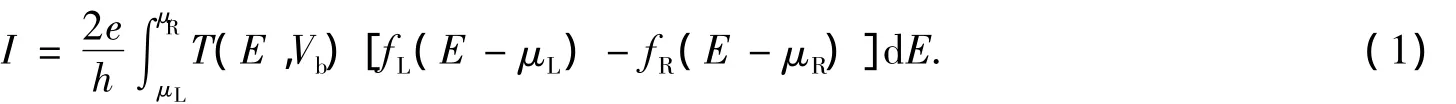
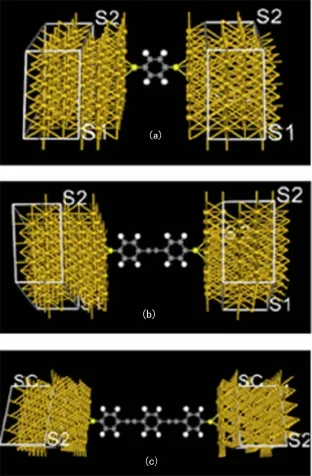
图1 含苯环巯基分子与Au电极组成的分子桥
式(1)中:μL,μR分别为左右电极的电化学势;Vb为两端偏压,与电化学势满足如下关系:

fL,fR分别是左右电极的电子费米分布函数

式(1)中的T(E,Vb)是能量为E,偏压为Vb时体系的透射系数,可用推迟和超前格林函数GR(E)和GA(E)表示为[21]
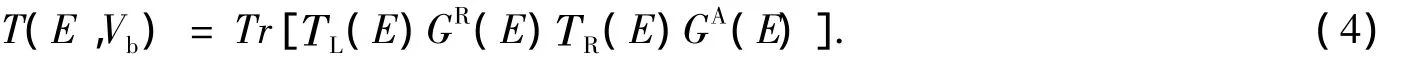
式(4)中,TL(E),TR(E)是左右电极的耦合矩阵,可用分子对散射区域
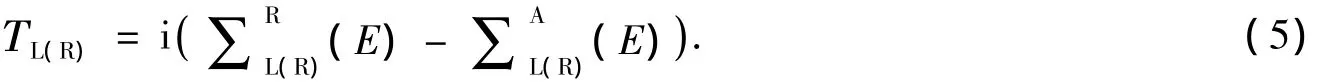
透射系数T(E)可以分解为n个本征能量通道

而系统平衡态的电导[22]可用费米能Ef处的透射系数T(Ef)表示

在计算中,透射谱及I-V特性的计算是采用基于密度泛函理论(DFT)和非平衡格林函数(NEGF)[23]的计算程序包Atomistix ToolKit(ATK)完成的.有机分子和Au电极都选DZP(doubleζ+polarization)为基矢.内层电子用Troullier-Martins赝势[16],截断能量mesh cut-off取200 Ry(约2 720 eV).电子交换关联势为广义梯度近似(GGA)[14,24].
2 计算结果与讨论
选取A,B,C分别含1个、2个和3个苯环的3种分子桥作为研究对象,通过比较来揭示透射率与苯环数的关系.当无外加偏压时(Vb=0),3种情况的透射率 T(E,Vb=0)见图2.由图2可知:含1个苯环的巯基分子(见图2中A)出现2个透射率几乎相一致的透射峰,能量分别为-1.0 eV和2.36 eV,2个峰之间的谷底较宽,约有1.0 eV;2个苯环的系统(见图2中B)出现了3个透射峰,对应的能量分别为-0.80,1.73,2.82 eV,在负能区的透射峰明显低于正能区的2个峰值,而且正能区的2个峰相当于一对双峰,几乎有相同的透射率;当增加到3个苯环时(见图2中C),透射峰的数目并没有增加,仍为3个峰,相应的能量分别为 -0.75,1.55,2.33 eV.因此,从图2不难看出,随着苯环数的增加,电子的透射率降低,透射峰变窄并且其值减小,透射峰的位置发生移动,都向着零能区靠拢.实际上,随着苯环数的增加,分子能级间距相对减小,LUMO和HOMO之间的能量差也减小.透射率的变化是分子轨道能级变化的直接反映.
图3是3个分子体系的I-V曲线.3个含苯环巯基分子体系(见图3中的A,B,C)都在电压小的范围出现电流禁区,然后开通,而A比B推迟约0.6 V,B又比C推迟约0.2 V(这一结果与文献[25]不同取代基NO2,Cl等情况相比稍有不同).开通后电流线性增加,呈欧姆性质,每个电流开通位置与第1透射峰位置分布相一致.接着电流大幅线性增加,欧姆性质改变与第2透射峰位置分布相一致,A明显比B和C的电输运性质有优势,C最差.
图4是体系在0~6 V偏压下的电导分布图.比较发现:3个含苯环巯基分子体系中(见图4中的A,B,C),电压比较低的情况下电导很小,电流也很小.分别在2.4,1.7,1.5 V左右出现第1电导平台,与第1透射峰和电流第1线性区间相对应,这说明未占轨道的导通为建立电导平台提供了条件.接着电压升高,伴随其他分子轨道的相继开通,各分子体系又开始出现更高的电导平台.A的电导平台最宽最高,B比C稍高稍宽.
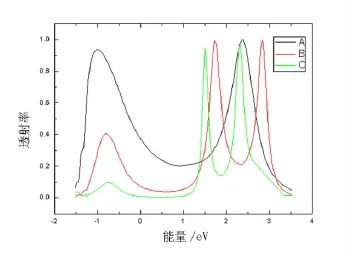
图2 无偏压时3种分子桥的透射率随能量的变化
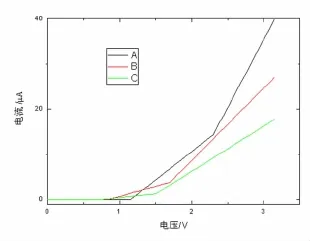
图3 3种含苯环巯基分子桥的I-V曲线
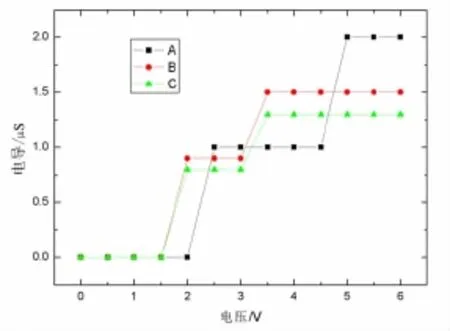
图4 分子桥电导与外加偏压的关系
研究结果表明,在非共振隧穿条件下,分子结的电阻与分子长度存在下面的关系[26]:

式(8)中:R0是扩展分子的电阻;R是有效接触电阻;s为分子的长度;β为衰减常数,即长度隧穿衰减因子,该方程称为非共振隧穿方程.由图4可以看出,当外加电压为1.55 V时,分子出现共振隧穿,因此计算衰减常数β时应用外加电压小于1.55 V时的电导.图5给出了在外加电压为0~1.55 V时线性拟合出来的长度隧穿衰减因子β值,其中横坐标为苯环的个数,纵坐标则是以e为底的分子的电阻.图5中3点分别为3个分子在外加电压为0~1.55 V的电阻自然对数值,而这里的电阻为这一偏压范围内的平均值.图5中的直线是通过对3点的线性拟合得到的,该直线的斜率即代表了衰减常数β值的大小,该直线与纵轴的截距即是有效接触电阻的大小.计算得到有效接触电阻大小约为2.9×108Ω,衰减常数 β =2.1 nm-1.实验[21]测量 β 值约为(1.7 ±0.1)nm-1,计算所得到的数值与实验测量所得到的数值符合.
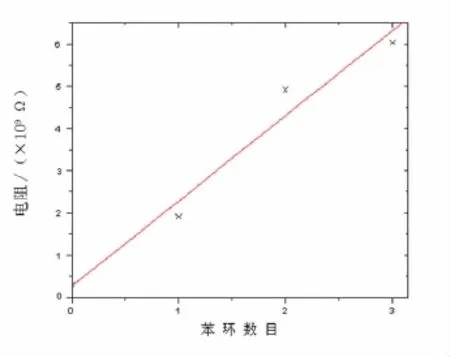
图5 苯环个数与电阻的关系
3 结论
本文从第一性原理出发,利用密度泛函和非平衡格林函数理论方法,研究了含不同数目苯环的巯基分子的透射谱、电流、电导等,比较了在不同的几何结构下分子的电输运性质.发现苯环数目增加,第1透射峰提前,但透射峰变窄小,计算发现该类分子的电导曲线都显示出良好的平台特性,苯环数目增加,电导平台变窄,导电能力变弱.分子长度是影响分子电输运特性的重要因素,通过计算得到长度隧穿衰减因子β值为2.1 nm-1,与实验结果很好地符合.
[1]Reed M A,Zhou C,Muller C J,et al.Conductance of a molecular junction[J].Science,1997,278(5336):252-253.
[2]Collins P G,Arnold M S,Avouris P.Engineering carbon nanotubes and nanotube circuits using electrical breakdown[J].Science,2001,292(5517):706-709.
[3]Park J,Pasupathy A N,Goldsmith J I,et al.Coulomb blockade and the Kondo effect in single-atom transistors[J].Nature,2002,417(13):722-725.
[4]Mujica V,Kemp M,Ratner M A.Electron conduction in molecular wires[J].J Chem Phys,1994,101(8):6849-6855.
[5]Ventra M D,Pantelides S T,Lang N D.First-principles calculation of transport properties of a molecular device[J].Phys Rev Lett,2000,84(5):979-982.
[6]Tian W,Datta S,Hong S,et al.Conductance spectra of molecular wires[J].J Chem Phys,1998,109(7):2874-2882.
[7]Li Z L,Zou B,Wang C K,et al.Electronic transport properties of molecular bipyridine junctions:Effects of isomer and contact structures[J].Phys Rev B,2006,73(7):075326.
[8]Wang C K,Luo Y.Current-voltage characteristics of single molecular junction:Dimensionality of metal contacts[J].J Chem Phys,2003,119(9):4923-4928.
[9]夏蔡娟,房常峰,胡贵超,等.官能团对分子器件电输运特性的影响[J].物理学报,2008,57(5):3148-3153.
[10]Keane Z K,Ciszek J W,Tour J M,et al.Three-terminal devices to examine single-molecule conductance switching[J].Nano Lett,2006,6(7):1518-1521.
[11]Chen J,Reed M A,Rawlett A M,et al.Large on-off ratios and negative differential resistance in a molecular electronic device[J].Science,1999,286(5336):252-253.
[12]Xia Caijuan,Fang Changfeng,Hu Guichao.Effects of contact atomic structure on electronic transport in molecular junction[J].Chin Phys Lett,2008,25(5):1840-1843.
[13]He J,Chen F,Li J,et al.Electronic decay constant of carotenoid polyenes from single-molecule measurements[J].J Am Chem Soc,2005,127(5):1384-1385.
[14]Ventra M D,Pantelides S T,Lang N D.First-principles calculation of transport properties of a molecular device[J].Phys Rev Lett,2000,84(5):97-98.
[15]夏蔡娟,房常峰,胡贵超,等.分子的位置取向对分子器件电输运特性的影响[J].物理学报,2007,56(8):4884-4890.
[16]Taylor J,Guo Hong,Wang Jian.Ab initio modeling of open systems:Charge transfer electron conduction and molecular switching of a C60 device[J].Phys Rev B,2001,63(12):121104.
[17]Haug H,Jauho A P.Quantum kinetics in transport and optics of semiconductors[M].Berlin:Springer,1996.
[18]Mehrez H,Wlasenko A,Larade B,et al.I-V characteristics and differential conductance fluctuations of Au nanowires[J].Phys Rev B,2002,65(19):195419.
[19]Reichert J,Weber H B,Mayor M,et al.Low-temperature conductance measurements on single molecules[J].App Phys Lett,2003,82(23):4137-4139.
[20]Geng H,Yin S,Chen K Q,et al.Effects of intermolecular interaction and molecule-electrode couplings on molecular electronic conductance[J].J Phys Chem B,2005,109(25):12304-12308.
[21]Datta S.Electronic transport in mesoscopic systems[M].Cambridge:Cambridge University,1995.
[22]Venkataraman L,Klare J E,Nuckolls C,et al.Dependence of single-molecule junction conductance on molecular conformation[J].Nature,2006,442:904-907.
[23]Brandbyge M,Mozos J L,Ordejon P,et al.Density functional method for non-equilibrium electron transport[J].Phys Rev B,2002,65(16):165401.
[24]Ventra M D,Kim S G,Pantelides S T,et al.Temperature effects on the transport properties of molecules[J].Phys Rev Lett,2001,86(2):288-291.
[25]Bauschlicher C W,Lawson J W.Current-voltage curves for molecular junctions:Effect of substitutients[J].Phys Rev B,2007,75(11):115406.
[26]Kim B S,Beebe J M,Jun Y,et al.Correlation between HOMO alignment and contact resistance in molecular junctions:Aromatic thiols versus aromatic isocyanides[J].J Am Chem Soc,2006,128(15):4970-4971.
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
高中数理化(2020年1期)2020-02-29 02:21:18
电子制作(2018年14期)2018-08-21 01:38:38
中国眼镜科技杂志(2018年13期)2018-08-11 06:06:10
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
电测与仪表(2016年20期)2016-04-11 11:37:46
西南医科大学学报(2016年4期)2016-01-03 01:26:33
激光与红外(2015年11期)2015-03-23 06:07:32
中国药业(2014年21期)2014-05-26 08:56:50
建筑材料学报(2014年6期)2014-03-11 17:08:59
