
乙烯基噻吩共轭螺噁嗪化合物的密度泛函理论研究
2011-12-11 09:09:54孙海涛田晓慧元以中孙金煜孙真荣卓小玲
物理化学学报 2011年8期
孙海涛 田晓慧,* 元以中 孙金煜 孙真荣 卓小玲
(1华东理工大学材料科学与工程学院,超细材料制备与应用教育部重点实验室,上海200237; 2华东师范大学,精密光谱科学与技术国家重点实验室,上海200062)
乙烯基噻吩共轭螺噁嗪化合物的密度泛函理论研究
孙海涛1田晓慧1,*元以中1孙金煜1孙真荣2卓小玲1
(1华东理工大学材料科学与工程学院,超细材料制备与应用教育部重点实验室,上海200237;2华东师范大学,精密光谱科学与技术国家重点实验室,上海200062)
采用密度泛函理论(DFT)方法,在B3LYP/6-31G*水平上对乙烯基噻吩共轭螺噁嗪化合物SO-SO3的几何构型、电子结构、前线分子轨道等进行了理论研究,计算结果表明:SO-SO3的开环过程会使得开环体的左右两个部分键长均等化,导致共轭体系变大,能隙明显减小;乙烯基噻吩基团共轭接入螺噁嗪母体后,导致体系的共轭作用变大,在激发态下电子流动增强,形成由乙烯基噻吩向萘并噁嗪的有效电荷转移与能量转移;结合前线分子轨道成分分析乙烯基噻吩单元在最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)中的轨道贡献率明显增加.含时密度泛函理论(TD-DFT)计算的电子吸收光谱结果显示:当接入的乙烯基噻吩单元达到2-3个时,影响SO2和SO3开环的最低能量激发态变为第一激发单重态S1,并且均源自电子从HOMO至LUMO的跃迁且为π-π*跃迁;其最大吸收波长λmax达到466-540 nm,且红移十分明显,其对应开环体O-SO2与O-SO3的λmax达到605和647 nm.
乙烯基噻吩;螺噁嗪;密度泛函理论;前线分子轨道;电子吸收光谱
1 引言
螺噁嗪化合物的光致变色现象首次被Fox1报道,他合成了第一个螺噁嗪化合物-1,3,-二氢-1,3,3-三甲基[2H]-吲哚螺-2,3ʹ-[3H]萘并[2,1-b][1,4]噁嗪(NISO),后来经证实此类化合物的光化学性质较偶氮类,螺吡喃类具有更高的光稳定性和抗疲劳性,有潜在的应用前景,因而近年来得到了广泛深入的研究.2-4与此同时,噻吩类化合物作为共轭电致变色材料的典型代表以其良好的稳定性、可加工性,独特的半导体特性和多样的可调节性质同样引起了人们很大的研究兴趣.2因此,将含噻吩单元通过共轭的方式引入到螺噁嗪母体中制成具有光致变色和光电转化的新型双功能材料,在拓展了两类材料应用的基础上希望能获得新的光学性质,并且在数据存储、显示材料、光电材料、生物分子活性的光调控、光计算以及防伪鉴伪方面具有潜在的应用前景.与传统的大量论文和专利报道的吲哚啉萘并螺噁嗪相比,对于噻吩共轭螺噁嗪类化合物的报道很少,理论研究也相对较少.随着密度泛函理论的发展与成熟,将其应用于许多有机化合物的理论计算已成为当前一个非常活跃的研究领域.5-7本文通过改变乙烯基噻吩共轭基团连接的长度设计了一系列新型的乙烯基噻吩共轭螺噁嗪衍生物SO-SO3 (图1),并通过密度泛函理论,研究了这类化合物的结构和性能的关系、电子结构、前线分子轨道以及吸收光谱性质等,为进一步设计和合成具有可见光致变色及光电转化的新型双功能的材料提供比较可靠的方向性引导和有力的理论依据.
2 计算方法
在量子化学计算中,要得到可靠的结果,计算模型和基组的合理选择是必要的.8,9考虑到计算资源和计算精度等因素,本文采用密度泛函DFT/ B3LYP10(a,b)方法,以6-31G*基组依次对乙烯基噻吩共轭螺噁嗪衍生物SO-SO3及其相应的开环体的几何构型进行全优化,分析了分子的结构特点及相互关系,计算得到了最高占据轨道(HOMO)、最低空轨道(LUMO)及能隙.采用自然键轨道(NBO)分析获得原子上自然电荷分布,通过组态相互作用(CIS)计算得到其基态和第一单激发态的电子结构.在优化得到的基态几何构型的基础上,采用含时密度泛函(TD-DFT)10(c,d)方法研究了其电子吸收光谱的性质.所有计算都利用Gaussian 09程序11并在Pentittm IV微机上完成,计算的收敛精度均采用程序内定值.
3 结果与讨论
3.1 几何构型的优化
如图1所示,吲哚啉螺噁嗪化合物的分子结构可分为两部分,左边是吲哚环部分,右边是萘并噁嗪部分,它们以sp3杂化的螺碳原子连接,这两部分基本上相互垂直,在紫外光的激发下,SO(spirooxazine)分子中的螺C―O键发生异裂,引起分子的结构以及电子的组态发生异构化和重排,通过螺碳原子连接的两个环系由正交变为共平面,螺碳原子由sp3杂化变为sp2杂化,同时形成了一个大的共轭体系.在可见光或热的作用下,开环体发生关环反应返回到SO,构成了一个典型的光致变色体系.2Schneider12(a)对螺噁嗪的开环过程进行了研究,经过CARS方法测定和PPP方法计算结果证实是各种异构体的混合物并且至少有四种异构体是可以相对稳定存在的,其中最为稳定的是反-反-顺(TTC)构型.12(b)为方便讨论及节省计算机时,文中仅以TTC构型作为开环体的代表进行理论计算.
结合图1和表113给出了SO-SO3及其开环体(O-SO)分子在B3LYP/6-31G*水平下优化的部分键长参数以及文献值,并且优化得到的键长与文献值的误差仅为1%左右,证实了计算所采用的泛函和基组对于本体系研究的可靠性.由表1可见, SO-SO3在变为开环体的过程中,其化学键1-6的键长发生了明显的变化,整体趋势为长键变短,短键变长,表明形成的开环体的左右两个部分具有键长均等化的趋势,键1-6键长的平均值由0.1400 nm减少至0.1361 nm,说明了开环形成了明显的离域化结构.此外,螺C―O键(键7)的键长随着连接的乙烯基噻吩单元的增加而具有略微增大的趋势,这使得在反应过程中键的断裂需要的能量更低,因此这将有利于在紫外辐射甚至可见光照射下螺C―O键的断裂和开环体的形成.表1还给出了乙烯基噻吩基团的碳碳单双键交替键长差值(BLA)值,14所谓BLA值就是指对于含有CC单双键交替的化合物或基团的C―C键长的平均值和C=C键长的平均值之差.由表1可见,SO1-SO3和对应开环体的乙烯基噻吩基团的BLA值逐渐减小,表明其共轭体系的扩大;其中开环体的乙烯基噻吩基团的BLA值比相应闭环体的略小,表明开环过程中电子云流动增强可以使螺噁嗪萘环部分与乙烯基噻吩基团共轭作用变大但是影响不大.此外计算所得SO-SO3的偶极矩由2.7819 C·m增加到4.8367 C·m,说明乙烯基噻吩的引入使得基态分子的极性增大.
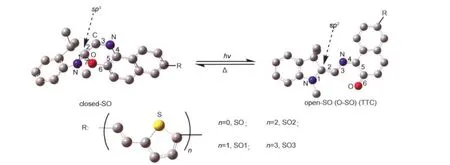
图1 SO-SO3分子结构模型及示意图Fig.1 Molecular structural model and sketch map of SO-SO3H atoms are omitted for clarity.
3.2 电子结构
采用NBO分析获得部分原子和基团上自然电荷分布.通过计算发现,在接入不同长度的乙烯基噻吩单元后吲哚N原子上的电荷(-0.470e)并不发生明显变化,但从化合物SO到SO3其噁嗪O原子上的电荷由-0.538e减小到-0.536e,说明在萘环上接入乙烯基噻吩基团后不影响吲哚环部分的电荷分布,但是噁嗪O上的电荷密度有减少的趋势,这样的电荷变化显然会破坏化合物的稳定性,使得螺C―O键的断裂容易,增大开环速率.同样比较SO和SO1的电荷密度剖面图也可以看出接入乙烯基噻吩基团后萘并噁嗪环的剖面线条更加均匀,电荷密度分布更均匀,说明共轭效应作用明显增强.
此外,分别计算了SO-SO3的吲哚环(含螺C原子)、萘并噁嗪、乙烯基噻吩单元三个部分的基态和第一激发单重态(开环前)的电荷分布(见图2).正号表示有电子流入;负号表示有电子流出.由图可见,分子在由基态到激发态过程中,含螺C吲哚环部分和乙烯基噻吩单元部分分别有大小不等的电子流向萘并噁嗪部分,其中,比较SO和SO1的萘并噁嗪部分的电荷,发现后者(+0.104e)几乎是前者(+0.051e)的两倍,随着乙烯基噻吩单元的增加,电荷变化逐渐变大,说明乙烯基噻吩基团可以显著增强螺噁嗪分子的萘并噁嗪部分的电子流动.
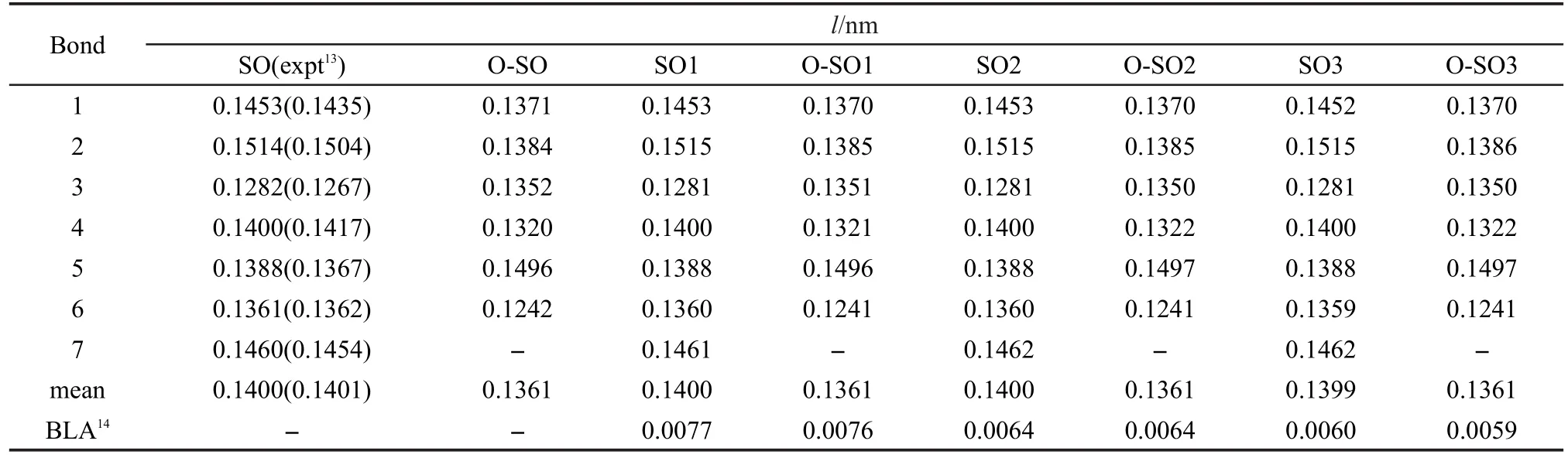
表1 闭环体SO-SO3及相应的开环体(O-SO)分子在B3LYP/6-31G*水平下优化的部分键长(l)和乙烯基噻吩基团的碳碳单双键交替键长差值(BLA)(nm)Table 1 Selected B3LYP/6-31G*skeletal bond lengths(l)for SO-SO3 and the corresponding open-forms(O-SO)and the mean single-double CC bond length alternation(BLA)values for vinyl thiophene group
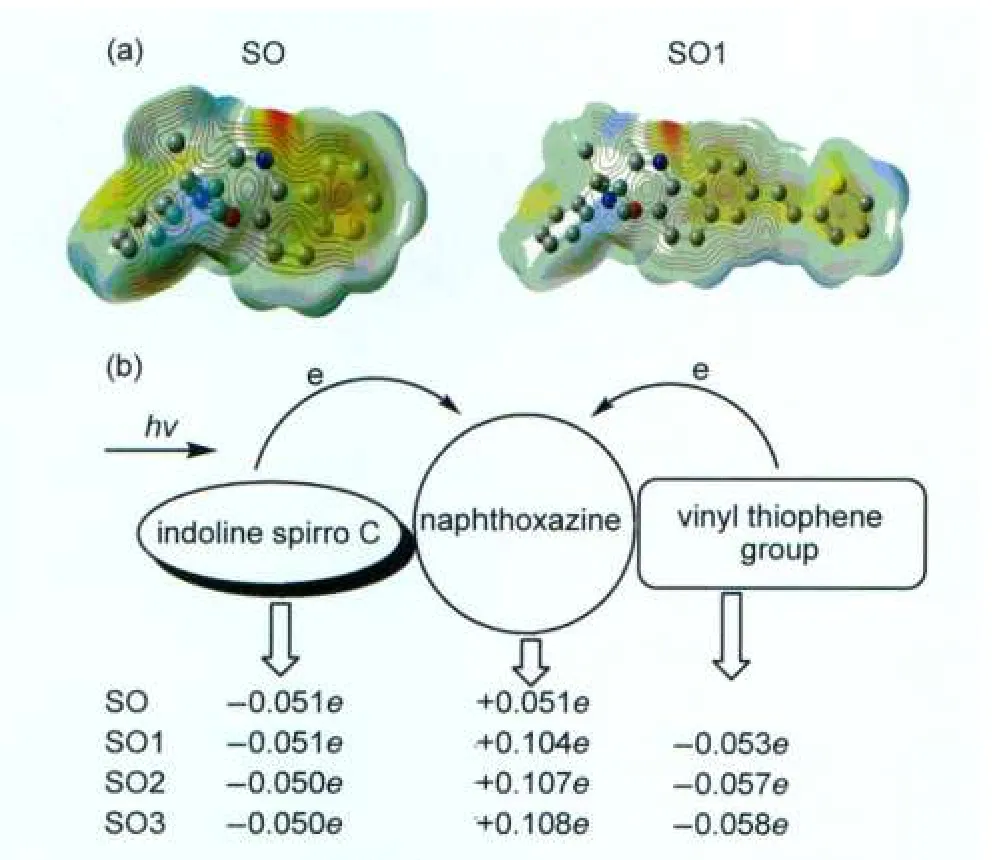
图2 (a)SO和SO1的电荷密度剖面图以及(b)SO-SO3的基态和第一单激发态的电荷分布Fig.2 (a)Profile diagram of charge density for SO and SO1 and(b)charge distribution on the ground and first single state of SO-SO3
3.3 前线分子轨道
HOMO和LUMO对分子性质的影响非常重要.15,16图3和图4分别给出了B3LYP/6-31G*计算所得SO-SO3及其对应开环体的前线分子轨道,即HOMO和LUMO的轨道及能量,图5给出了其各个部分与整体态密度分布状况,图6给出了SO-SO3以及其开环体的能隙变化.
根据前线轨道理论,螺噁嗪的分子轨道可以由吲哚环和萘并噁嗪两部分相互作用构成.由图3和图5可见,SO的HOMO主要分布在吲哚环(占77%)而LUMO主要分布在萘并噁嗪部分(占96%),在SO中,由于轨道分布在由螺碳原子连接的两个不同的部分因此并没有发生明显的作用,其HOMOLUMO的跃迁主要对应于从吲哚环到萘并噁嗪的电荷转移跃迁,基于前线轨道对称性,这种跃迁的强度很低.当萘环上连接不同长度的乙烯基噻吩基团后,SO1-SO3分子的前线轨道较SO发生变化即HOMO和LUMO基本都分布在除吲哚环外的π共轭骨架上(由萘并噁嗪与乙烯基噻吩两部分组成),并分别显示出典型的π和π*轨道特征.乙烯基噻吩单元在HOMO和LUMO中的轨道贡献率分别由45%和31%增加到86%和90%,这说明由于乙烯基噻吩本身强的π电子的离域性,改变了螺噁嗪母体电子云分布以及前线分子轨道的分布.
同时,能隙在逐渐减小,说明乙烯基噻吩的引入增加了其与萘并噁嗪部分的共轭作用;其中, HOMO能均增大,体系易失去电子,即易产生空穴; LUMO能均减小,表明体系易接受电子,电子注入能力提高了,因此使得电子云流动变得更加容易.由图6可以发现,开环体的能隙较未开环的明显减小,这主要是开环过程分子结构形成了一个更大的π共轭体系造成的(由图4的HOMO轨道分布可见).
3.4 吸收光谱

图3 B3LYP/6-31G*计算所得SO-SO3的前线分子轨道及能隙(eV)Fig.3 Frontier molecular orbitals and energy gap(eV)of SO-SO3 calculated using B3LYP/6-31G*

图4 B3LYP/6-31G*计算所得SO-SO3对应的开环体的前线分子轨道及能隙(eV)Fig.4 Frontier molecular orbitals and energy gap(eV)of the corresponding open-forms of SO-SO3 calculated using B3LYP/6-31G*
用含时密度泛函TD-B3LYP/6-31G*计算得到SO-SO3及其开环体的电子吸收光谱,最低三个单重激发态(S1-S3)及其对应的吸收波长λ,振子强度f以及跃迁类型.由表2可见,对于SO分子,TDDFT的计算预测与文献10(c,d)实验值显示了良好的的一致性,说明了此计算方法的可行性.为了讨论不同基组下对于TDDFT计算电子光谱的影响,分别在6-31G*,6-31+G*,以及6-311+G*下对SO分子进行了TD-DFT的电子光谱计算,由表2可以发现基组增加弥散函数后,吸收波长变化不大,相对于基组6-31G*仅发生4-6 nm的红移,说明对于本体系在TD-B3LYP/6-31G*下达到计算精度和计算效率的最优化.但计算所得结果与实验结果略有偏差,造成计算结果与实验值偏差的原因是计算方法和实验方法都有各自的缺陷,比如计算中都需要引入一些近似,计算值所处的环境是在真空态下,但因理论计算结果中考虑了必要的相关效应,保留了重要因素的作用,对于实验预测仍是可信的.
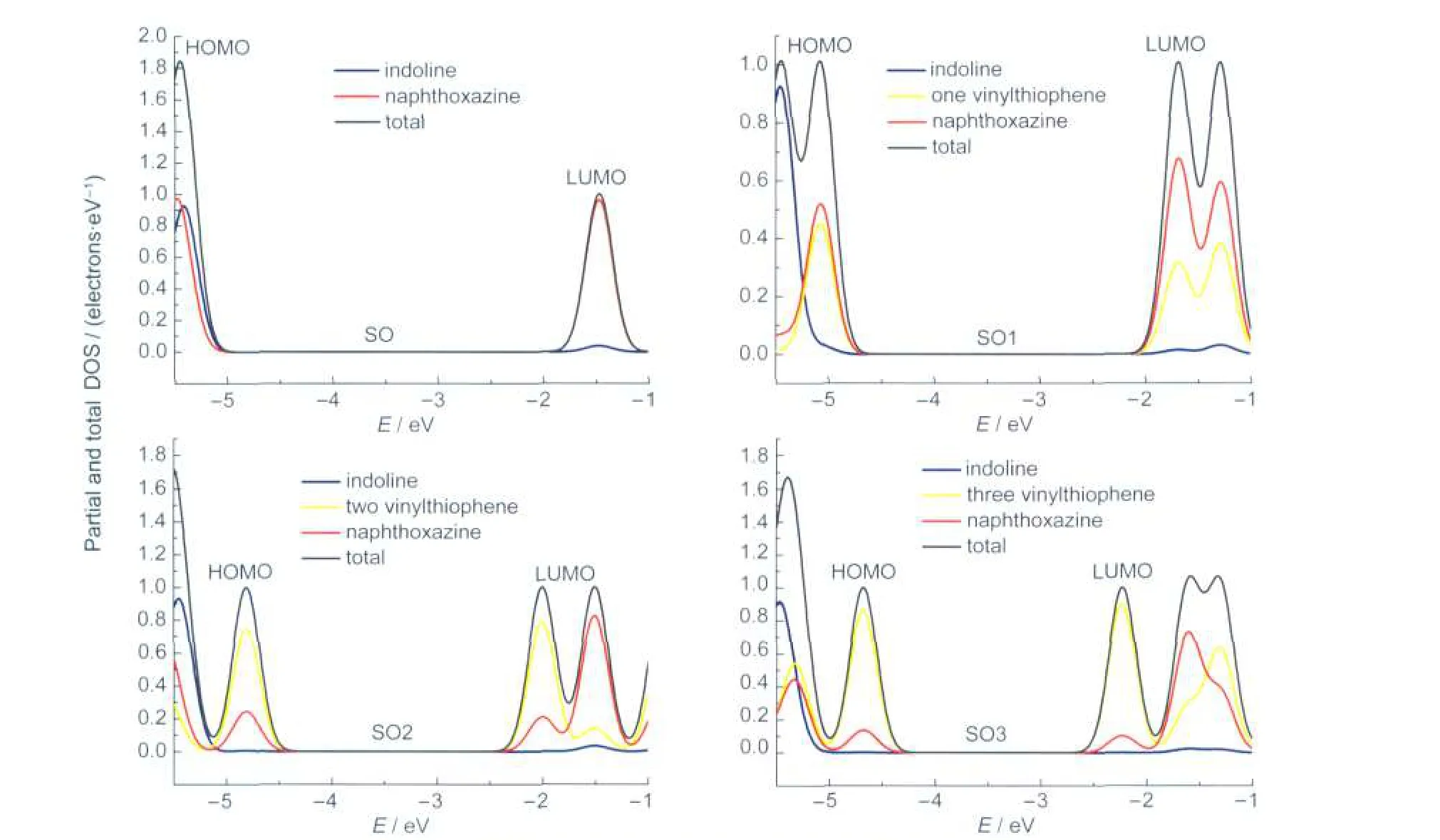
图5 SO-SO3的各个部分与整体态密度(DOS)分布图Fig.5 Diagrams of partial and total density of states(DOS)for SO-SO3
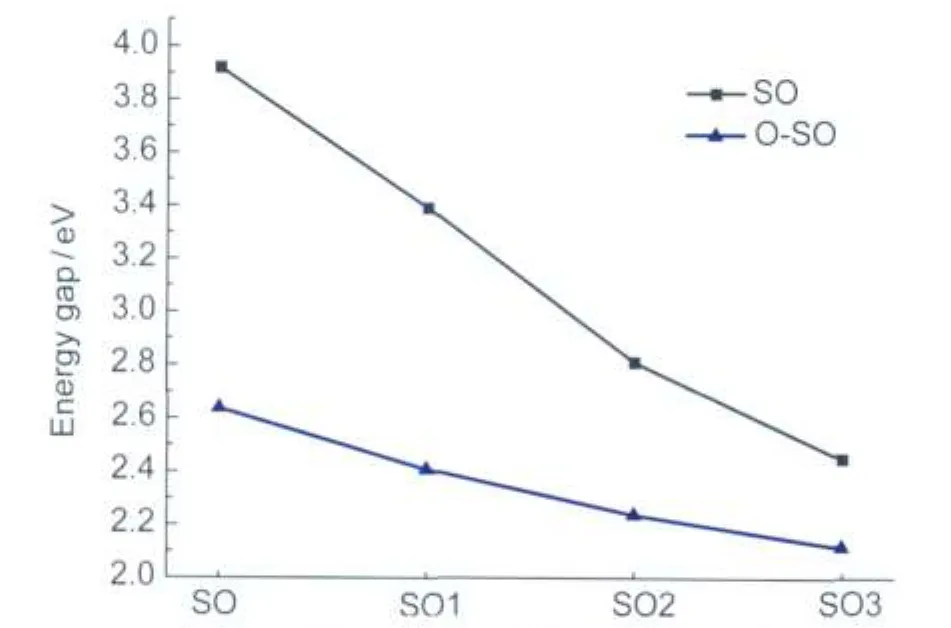
图6 SO-SO3及其对应开环体的能隙Fig.6 Energy gap of SO-SO3 and the corresponding open-forms
文献17报道螺噁嗪类化合物开环体的形成只经过激发单重态形成的.18由表2可知,SO的HOMO和LUMO分布在不同的环上,因此其电子跃迁相对具有较弱的振子强度(f=0.044),同时影响其最大吸收波长的最低能量激发态发生在第二单重态S2.而SO2和SO3的最大吸收波长发生在最低单重激发态S1,S1均源自电子从HOMO-LUMO的跃迁且为π-π*跃迁,并且分别具有较强的振子强度(f=1.910和f= 2.758),这说明随着乙烯基噻吩单元的增加,开环所需激发能降低,将更容易发生激发开环.
由图7可见,计算所得SO-SO3的最大吸收波长分别为344、364、466、540nm,可见由于乙烯基噻吩与螺噁嗪萘环部分的共轭作用,使得整体的吸收发生了显著的红移,特别是摩尔消光系数ε由103增大到105,在此位置的吸收强度明显增大.值得注意的是SO-SO3的最大吸收波长由紫外激发区转移到可见光激发区.此外,在同样的计算方法和基组下计算研究SO-SO3对应的开环体的电子吸收光谱(表3),对于O-SO而言,其最低单重激发态S1为σ→π*(LUMO)跃迁,其中σ轨道主要来源于氧原子和氮原子上的孤对电子并分布在噁嗪环上,根据轨道对称性,这种跃迁是禁阻的且其振子强度几乎可以忽略.而影响O-SO最大吸收的S0-S2跃迁表现为π-π*特征,且主要来源于电子从HOMO到LUMO的激发跃迁,由图4可见其HOMO和LUMO的轨道均分布在整个共轭平面结构上.对于O-SO2和O-SO3,其最大吸收波长发生在最低单重激发态S1,均源自电子从HOMO到LUMO的跃迁且同样呈现为典型π-π*跃迁.此外,从表3可以发现开环体吸收波长的变化趋势与闭环体类似,随着乙烯基噻吩单元的增加,从O-SO到O-SO3其吸收波长发生约68-170 nm的红移,其中O-SO2与O-SO3的最大吸收波长达到605和647 nm,相应的开环体理论上会在可见光区产生不同的颜色.由于接入2-3个乙烯基噻吩单元使得能够激发闭环体开环的最大吸收波长转移到可见光区,同时相应开环体在可见光区呈现出颜色,这就为设计和合成新型可见光致变色材料提供了可靠的理论依据,并且将会极大地拓展这类光学材料的应用范围.

表2 TD-B3LYP计算所得SO-SO3的电子吸收光谱,最低三个单重激发态及其对应的吸收波长λ、振子强度f以及跃迁类型Table 2 Electronic absorption spectra,wavelengths λ,oscillator strengths f,and the lowest three singlet excited states of SO-SO3 calculated with TD-B3LYP

图7 TD-B3LYP/6-31G*计算所得的SO-SO3的电子吸收光谱Fig.7 Electronic absorption spectra of SO-SO3 calculated with TD-B3LYP/6-31G*
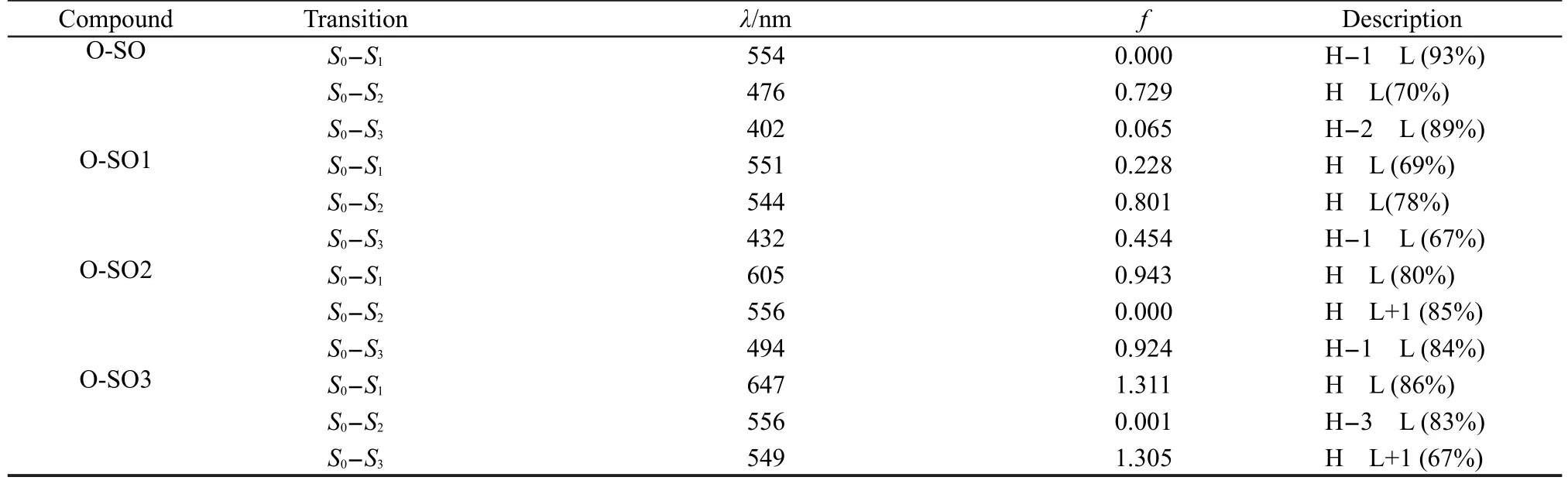
表3 TD-B3LYP/6-31G*计算所得SO-SO3对应的开环体的电子吸收光谱,最低三个单重激发态及其对应的吸收波长λ、振子强度f以及跃迁类型Table 3 Electronic absorption spectra,wavelengths λ,oscillator strengths f,the lowest three singlet excited states of the corresponding open-forms of SO-SO3 calculated with TD-B3LYP/6-31G*
4 结论
采用密度泛函理论和含时密度泛函方法,分别在B3LYP/6-31G*水平下研究和预测了乙烯基噻吩共轭螺噁嗪衍生物的几何构型、电子结构和电子吸收光谱并得到了较为理想的结果:(1)理论计算预测的结果和文献已有的实验值吻合较好,说明了理论计算的可靠性.(2)SO-SO3的开环过程会使得其共轭体系变大,带隙明显减小,最大吸收波长红移.(3)乙烯基噻吩基团共轭接入吲哚啉螺萘并噁嗪母体,由于乙烯基噻吩本身强的π电子的离域性,使得乙烯基噻吩与萘并噁嗪部分共轭作用增大;改变了萘并噁嗪部分的电子云分布同时使电子流动更容易,增加了整个分子的极性,降低了分子的带隙,改变了前线分子轨道分布特别是乙烯基噻吩单元在HOMO和LUMO中的轨道贡献率明显增加;当接入的乙烯基噻吩单元达到2-3个时,SO2和SO3的最低激发单重态(S1)均源自电子从HOMO到LUMO的跃迁,且为π-π*跃迁;影响最大吸收波长的最低能量激发态变为S1,激发开环将变得容易.最大吸收波长λmax达466-540 nm,红移十分明显并出现在可见光区,对光子的吸收效率显著增加,使得激发光源从紫外光向可见光区域发生转变,其中O-SO2与O-SO3的最大吸收波长达到605和647 nm,开环体会在可见光区产生相应的颜色变化(如从绿色到蓝色).这为设计和合成新型可见光致变色及光电转化材料提供了可靠的理论依据,将会极大地拓展这类光学材料的应用范围.此外,了解化学结构和电子吸收光谱等性质是对具有优良性能的材料进行分子结构设计和指导实验合成的起点,在DFT的理论计算框架下尤其是含时密度泛函的应用进行研究显示了与实验结果的规律的一致性,将为今后改构设计分子提供可靠的参考依据.19-21
(1) Fox,R.E.Research Reports and Test Items Pertaining to Eye Protection ofAir Crew Personnel.USAPatentAD440226[P]. 1961 Final Report on ContractAF41(657)215,AD 440226, 1961.
(2) Fan,M.G.Basic Fundamentals of Photochemistry and Materials Science of Photonics;Science Press:Beijing,2001; p 226.[樊美公.光化学基本原理与光子学材料科学.北京:科学出版社,2001:226.]
(3) Tamai,N.;Miyasaka,H.Chem.Rev.2000,100,1875.
(4) Chu,N.Y.C.Can.J.Chem.1983,61,300.
(5) Tang,Z.Y.;Hu,Y.C.;Zhao,Y.;Liu,S.B.Acta Phys.-Chim. Sin.2009,25,701.[唐智勇,胡云楚,赵 莹,刘述斌.物理化学学报,2009,25,701.]
(6) Xue,Y.S.;Gong,X.D.;Xiao,H.M.;Tian,H.Acta Chim.Sin. 2004,62,963.[薛运生,贡雪东,肖鹤鸣,田 禾.化学学报, 2004,62,963.]
(7) Wang,J.Y.;Feng,C.G.Chinese Journal of Applied Chemistry 2007,24,7. [王建营,冯长根.应用化学,2007,24,7.]
(8) Kanis,D.R.;Ratner,M.A.;Marks,T.J.Chem.Rev.1994,94, 195.
(9) Dehu,C.;Meyers,F.;Bredas,J.L.J.Am.Chem.Soc.1993,115, 6198.
(10) (a)Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785. (b)Becke,A.D.J.Chem.Phys.1993,98,5648. (c)Maurel,F.;Aubard,J.;Millie,P.;Dognon,J.P.;Rajzmann, M.;Guglielmetti,R.;Samat,A.J.Phys.Chem.A 2006,110, 4759. (d)Sheng,Y.H.;Leszczynski,J.;Garcia,A.A.;Rosario,R.; Gust,D.;Springer,J.J.Phys.Chem.B 2004,108,16233.
(11) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision B.01;Gaussian Inc.:Pittsburgh,PA,2010.
(12) (a)Schneider,S.Z.Phys.Chem.Neue.Folge.1987,154,91. (b)Maeda,S.;Mitsuhashi,K.;Osano,Y.T.;Nakamura,S.;Ito, M.Mol.Cryst.Liq.Cryst.1994,246,223.
(13) Millini,R.;Del Piero,G.;Allegrini,P.;Crisci,L.;Malatesta,V. Acta Crystallogr.C 1991,47,2567.
(14) Ortiz,R.P.;Delgado,M.C.R.;Casado,J.;Hernández,V.;Kim, O.;Woo,H.Y.;Navarrete,J.T.L.J.Am.Chem.Soc.2004,126, 13363.
(15) Cufioni,A.;Andreoni,W.IBM J.Res.Dev.2001,45,101.
(16) Cufioni,A.;Mauro,B.;Andreoni,W.Chemical Physics Letters 1998,294,263.
(17) Bohne,C.;Fan,M.G.;Li,Z.J.;Lusztyk,J.;Scaiano,J.C. J.Chem.Soc.Chem.Commun.1990,571.
(18) (a)Tyer,N.W.,Jr.;Becker,R.S.J.Am.Chem.Soc.1970,92, 1289. (b)Tyer,N.W.,Jr.;Becker,R.S.J.Am.Chem.Soc.1970,92, 1295.
(19)Wang,X.W.;Jiang,G.;Du,J.G.Acta Phys.-Chim.Sin.2011, 27,309.[王晓巍,蒋 刚,杜际广.物理化学学报,2011, 27,309.]
(20) Kumar,S.;Velasco,K.;McCurdy,A.J.Mol.Struct.2010,968, 13.
(21) Hudson,G.A.;Cheng,L.;Yu,J.M.;Yan,Y.;Dyer,D.J.; McCarroll,M.E.;Wang,L.C.J.Phys.Chem.B 2010,114,870.
April 21,2011;Revised:May 18,2011;Published on Web:July 1,2011.
Density Functional Theory Study on Vinyl Thiophene Group Conjugated Spirooxazines
SUN Hai-Tao1TIAN Xiao-Hui1,*YUAN Yi-Zhong1SUN Jin-Yu1SUN Zhen-Rong2ZHUO Xiao-Ling1
(1Key Laboratory for Ultrafine Materials of Ministry of Education,School of Materials Science and Engineering,East China University of Science and Technology,Shanghai 200237,P.R.China;2State Key Laboratory of Precision Spectroscopy, East China Normal University,Shanghai 200062,P.R.China)
We carried out a theoretical study on the geometries,electronic structures,and frontier molecular orbitals of vinyl thiophene group conjugated spirooxazines(SO-SO3)using density functional theory(DFT)at the B3LYP/6-31G*level.The calculated results show that the equalization of bond lengths at the left and right parts of the open-forms occurred during the ring-opening process.A large conjugated system was formed and this significantly narrowed the energy gap.The conjugated system became larger and its electrons flowed easily because of the introduction of different lengths of vinyl thiophene conjugation moieties into the spirooxazine molecule.The electrons and energy efficiently transferred from the vinyl thiophene to naphthoxazine.The orbital contribution rate of the vinyl thiophene group in the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO) increased obviously.Time-dependent DFT(TD-DFT)calculations showed that as the conjugated vinyl thiophene unit reached 2-3 the first singlet excited state of SO2 and SO3 resulted from the electron transition from the HOMO to the LUMO,which were also assigned to the π-π*transition.Meanwhile,λmaxwas between 466 and 540 nm with an obvious red-shift while the λmaxof O-SO2 and O-SO3 reached 605 and 647 nm,respectively.
Vinyl thiophene;Spirooxazine;Density functional theory;Frontier molecular orbital; Electronic absorption spectrum
∗Corresponding author.Email:tianxh@263.net;Tel:+86-21-64252167.
The project was supported by the National High Technology Research and Development Program of China(0099AA03500),Shanghai Leading Academic Discipline Project(B502)and Shanghai Key Laboratory Project,China(08DZ2230500,09JC1404300).
国家高技术研究发展计划(0099AA03500),上海市重点学科(B502)和重点实验室(08DZ2230500,09JC1404300)资助项目
O641
猜你喜欢
纤维复合材料(2018年2期)2018-12-07 00:41:24
电子测试(2018年1期)2018-04-18 11:52:24
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年4期)2016-01-17 09:01:04
合成化学(2015年10期)2016-01-17 08:56:47
石油化工建设(2015年4期)2015-12-01 04:17:09
海军航空大学学报(2015年1期)2015-11-11 17:22:41
橡胶工业(2015年6期)2015-07-29 09:20:38
应用化工(2014年9期)2014-08-10 14:05:08
原子与分子物理学报(2014年4期)2014-02-28 22:18:44

- 物理化学学报的其它文章
- Micellization Behavior of an Amphiphilic Drug Promethazine Hydrochloride-Surfactant System in an Aqueous Medium
- Synthesis of a Novel Thiadiazine Derivative and Electrochemical Properties for Pb2+Transfer across Water/1,2-Dichloroethane Interface
- YLuAG:Ce粉体的发光及闪烁特征:制备方法及缺陷效应
- 一种可作为FCC基质的新型改性镁铝尖晶石材料
- 纳米碳纤维载铂作为质子交换膜燃料电池阳极催化剂
- 用于单分子动力学实验的微流控混合器