
N-氰基-亚胺酯衍生物的合成新方法*
2011-11-27 04:45:14马文博
合成化学 2011年4期
马文博,尹 平,何 菱
(四川大学 华西药学院 靶向药物与释药系统教育部重点实验室,四川 成都 610041)
N-氰基-亚胺酯(1)是重要的药物合成中间体[1,2],其合成方法文献报道不多。Robert Huffman等[3]采用原羧酸酯,单氰胺和乙酸酐于130 ℃~150 ℃反应制得1,收率中等。该方法的缺点是,若芳基底物连有取代基团时,则目标产物收率不足30%;由于原羧酸酯的制备需要使用毒性较大的氰基化合物,加上苛刻的高温条件,使其应用受到了限制。近年来有一些专利报道了1的合成方法,但内容都仅局限于在原路线下的工艺改进[4]。到目前为止,还没有采取芳基乙烯类化合物为原料合成1的相关报道。
本文报道合成1的新方法:以甲醇为溶剂,N-溴代丁二酰亚胺(NBS)为氧化剂,单氰氨与β-硝基芳基乙烯(2a~2e)反应合成了一系列N-氰基-亚胺酯衍生物(1a~1e,Scheme 1),其结构经1H NMR,13C NMR和HR-MS确证。该方法制备过程简易,反应条件温和,为进一步合成具有活性的杂环化合物合成提供了技术基础。此外,该反应实现了碳碳双键的断裂和胺化的串联反应,对于今后的该类方法学研究具有一定的借鉴意义。
在最初的合成设想中,我们拟采用单氰胺为氮源,以氮宾的插入方式与β-取代的苯乙烯反应合成氮杂环丙烷,但没有成功。
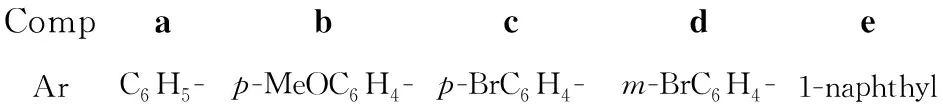
CompabcdeArC6H5-p-MeOC6H4-p-BrC6H4-m-BrC6H4-1-naphthyl
Scheme1
1 实验部分
1.1 仪器与试剂
Varian Inova 400型核磁共振仪(CDCl3为溶剂,TMS为内标 );Bruker Daltonics Data Analysis 3.2型质谱仪。
2a~2e,NBS,tBuONa,分析纯,成都科龙试剂有限公司;单氰胺,分析纯,北京百灵威试剂有限公司。
1.2 合成(以1a为例)
将β-硝基苯乙烯(2a)74.6 mg(0.5 mmol),单氰胺84.0 mg(2 mmol)和叔丁醇钠192.2 mg(2 mmol)溶于甲醇(2 mL)中,于室温搅拌30 min;加入NBS 356.0 mg(2 mmol),于50 ℃反应12 h。减压蒸除甲醇,残余物加水10 mL,用二氯甲烷(3×5 mL)萃取,合并萃取液,依次用饱和硫代硫酸钠溶液和饱和食盐水洗涤,无水Na2SO4干燥,浓缩后经硅胶柱层析[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=98∶2]分离得N-氰基-苯基亚胺甲酯(1a),产率51%。
用类似的方法合成1b~1e(洗脱剂:A=97∶3)。
1a: 无色液体,产率51%;1H NMRδ: 8.10~8.08(d,J=8.4 Hz, 2H), 7.65~7.61(t,J=7.6 Hz, 1H), 7.54~7.50(t,J=7.6 Hz, 2H), 4.07(s, 3H);13C NMRδ: 175.0, 133.9, 129.5, 129.0, 128.8, 113.5, 57.0; HR-MS(ESI)m/z: Calcd for C9H8N2ONa{[M+Na]+} 183.052 9, found 183.052 4。
1b: 白色固体,产率42%;1H NMRδ: 8.17~8.15(d,J=8.8 Hz, 2H), 6.99~6.97(d,J=8.8 Hz, 2H), 4.03(s, 3H), 3.89(s, 3H);13C NMRδ: 173.7, 163.9, 131.1, 121.6, 114.1, 113.9, 56.5, 55.6; HR-MS(ESI)m/z: Calcd for C10H10NO2Na{[M+Na]+} 213.063 4,found 213.064 7。
1c: 白色固体,产率43%;1H NMRδ: 7.99~7.97(d,J=8.4 Hz, 2H), 7.68~7.66(d,J=8.4 Hz, 2H), 4.06(s, 3H);13C NMRδ: 173.7, 132.3, 130.1, 129.0, 128.1, 112.9, 56.9; HR-MS(ESI)m/z: Calcd for C9H7N2ONa{[M+Na]+} 260.963 4, found 260.963 4。
1d: 白色固体,产率46%;1H NMRδ: 8.14~8.11(m, 2H), 7.77~7.75(m, 1H), 7.43~7.39(t,J=8.0 Hz, 1H), 4.08(s, 3H);13C NMRδ: 173.2, 136.7, 131.5, 131.0, 130.4, 127.3, 122.9, 112.7, 57.1; HR-MS(ESI)m/z: Calcd for C9H7N2ONa{[M+Na]+} 260.963 4,found 260.963 4。
1e: 白色固体,产率42%;1H NMRδ: 8.24~8.22(d,J=8.0 Hz, 1H), 8.10~8.09(d,J=6.8 Hz, 1H), 7.88(s, 2H), 7.68(s, 3H), 4.20(s, 3H);13C NMRδ:180.3, 133.4, 132.8, 129.2, 128.8, 128.6, 128.5, 127.6, 127.5, 125.5, 124.7, 113.6, 58.2; HR-MS(ESI)m/z: Calcd for C13H10N2ONa{[M+Na]+} 233.068 5,found 233.069 2。
2 结果与讨论
2.1 反应机理
在由2制取1的过程中,涉及到碳碳双键的断裂和胺化反应。我们认为首先在强碱和氧化剂的条件下,双键发生了断裂,继而通过缩合和甲醇加成得到氮杂缩醛中间体,最后通过NBS氧化脱氢得到1(Scheme 2)。本文使用tBuONa/NBS体系完成了碳碳双键的断裂,相对于其它断键方法相对较为温和;同时NBS的氧化条件也较为简易,为氮杂缩醛类衍生物的氧化方法提供了借鉴。
Scheme2
2.2 β-位取代基对产率的影响
以合成1a为模板,考察β-位取代基对1a产率的影响,结果见表1。由表1可见,当β-位取代基由NO2换为H时,无法得到1a;换为拉电子基(如氰基和甲酰基等)时,会得到相应的产物,但总体比较来看,还是β-位取代基为NO2时的产率较高。
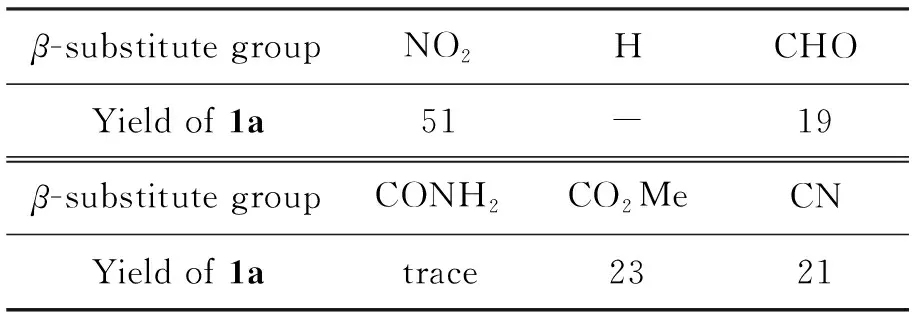
表1 β-位取代基对1a产率的影响*
[1] Zarguil A, Boukhris S, El Efrit M Letal.Easy access to triazoles,triazolopyrimidines,benzimidazoles and imidazoles from imidates[J].Tetrahedron Lett,2008,49:5883-5886.
[2] Huffman K R, Schaefer F C.N-cyanoimidates[J].J Org Chem,1963,28:1816-1821.
[3] He R J, Ching S M, Lam Y L. Traceless solid-phase synthesis of substituted xanthines[J].J Comb Chem,2006,8:923-928.
[4] Kammoun M, Chihi A, Hajjem B,etal.Facile and convenient synthesis of 1-perfuoroalkyl-1,2,4-triazoles[J].Synthetic Communications,2008,38:148-153.
猜你喜欢
油气·石油与天然气科学(2021年12期)2021-12-11 01:43:23
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
洛阳理工学院学报(自然科学版)(2020年1期)2020-05-15 09:24:02
四川警察学院学报(2019年6期)2019-12-28 07:20:06
分析化学(2017年12期)2017-12-25 12:43:03
文化产业(2016年6期)2016-10-19 19:13:47
合成化学(2015年2期)2016-01-17 09:04:21
化工进展(2015年6期)2015-11-13 00:27:23
中国塑料(2015年10期)2015-10-14 01:13:13
杭州师范大学学报(自然科学版)(2015年5期)2015-03-20 01:13:42
