
内-9-甲基-9-氮杂二环[3.3.1]壬-3-烷胺的合成工艺改进*
2011-11-26 01:50李顺来张汝涛杜洪光
合成化学 2011年1期
李顺来, 张汝涛, 王 军, 杜洪光
(北京化工大学 理学院,北京 100029)
盐酸格拉司琼是一种强效高选择性外周和中枢神经系统5-HT3受体拮抗剂, 直接作用于中枢化学感受区及外周迷走神经末梢的5-HT3受体, 抑制恶心和呕吐的发生, 对因放疗、化疗及手术引起的恶心和呕吐具有良好的预防和治疗作用[1~4]。
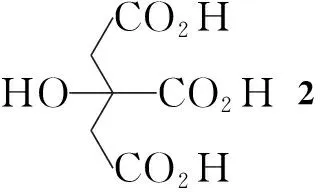
内-9-甲基-9-氮杂二环[3.3.1]壬-3-烷胺(endo-1)是制备盐酸格拉司琼的关键中间体,其合成方法[5~9]报道较少,本文在这些方法的基础上进行了探讨与改进。以柠檬酸(2)为起始原料,经浓硫酸-发烟硫酸混合液氧化脱羧制得丙酮二羧酸(3); 3与甲胺、戊二醛发生Mannich反应生成9-甲基-9-氮杂二环[3.3.1]壬-3-酮(4,假石榴皮碱);4与羟胺成肟得到9-甲基-9-氮杂二环[3.3.1]壬-3-酮肟(5, 3α-高托品酮肟);5采用变性LiAlH4还原合成了9-甲基-9-氮杂二环[3.3.1]壬-3-胺的内外异构体混合物(endo,exo-1); 再经柱层析成功分离出了endo-1(Scheme 1),总收率16.8%。制备endo-1的柱分离方法属首次报道。
该法操作步骤简单、反应条件温和、原料便宜,具有较好的实用性。
1 实验部分
1.1 仪器与试剂
XT4A型显微熔点仪(温度未校正);日本电子JNM-ECA 300 300 MHz型核磁共振谱仪(CDCl3为溶剂,TMS为内标);Bruker Vector 22型红外光谱仪(KBr压片);VG70-SE型气相色谱-质谱联用仪(LC-MS)。

Scheme1
所用试剂均为市售分析纯。
1.2 合成
(1)3的合成
冰盐浴冷却,在四口烧瓶中加入混酸[V(98% H2SO4) ∶V(20%发烟H2SO4)=3∶1] 150 mL,搅拌下于-5 ℃~-10 ℃慢慢加入2 76.8 g(400 mmol)(保持在0 ℃左右),待其完全溶解后再缓慢升温至放出大量气泡,升温至25 ℃(没有气泡放出为止)。用冰盐浴冷却至-5 ℃~-10 ℃后加入冰水中搅拌析晶,冷却,抽滤,滤饼用冰的乙酸乙酯洗涤,真空干燥得白色固体348.1 g,产率82.3%(70.5%[5],69.8%[6],90.0%[12]), m.p.135 ℃~136 ℃(138 ℃[12]); IRδ: 3 400~3 137(OH), 1 738(C=O), 1 700(C=O), 1 275(C-O), 1 442(O-H) cm-1。
(2)4的合成
在反应瓶中加入50%戊二醛35 mL,330 g(205 mmol),甲胺盐酸盐12 g和磷酸氢二钠21.6 g的水(320 mL)溶液,搅拌下于室温反应48 h;加入浓盐酸24 mL,于70 ℃反应2 h。冷却至室温,用10 mol·L-1NaOH溶液调至pH 10~11(出现黄色沉淀,颜色逐渐变深),用二氯甲烷(3×200 mL)萃取,合并萃取液,用饱和食盐水(80 mL)洗涤,无水硫酸钠干燥,减压蒸除二氯甲烷,残余物用乙醚-正己烷重结晶得淡黄色粉末412.0 g,收率38.2%(37.2%[5], 31.0%[7]), m.p.45 ℃~46 ℃(45 ℃~46 ℃[11]),Rf=0.18[展开剂:V(氯仿) ∶V(乙酸乙酯)=5 ∶1]; IRν: 1 706(C=O), 1 374(-CH3), 2 870(-CH3), 2 920(-CH2) cm-1; ESI-MSm/z: 153.9{[M+H]+}。
(3)5的合成
在反应瓶中加入醋酸钠15.3 g,盐酸羟胺14 g和乙醇80 mL,搅拌下滴加415.3 g(100 mmol)的乙醇(60 mL)溶液,回流反应4 h。浓缩至干,加水(55 mL)溶解,加10 mol·L-1氢氧化钠溶液35 mL,用二氯甲烷(3×150 mL)萃取,合并萃取液,用饱和食盐水(120 mL)洗涤,无水硫酸钠干燥,减压蒸除二氯甲烷,残余物用乙酸乙酯重结晶得黄色固体512.3 g,收率73.0%(72.4%[5], 31.0%[7]), m.p.129 ℃~130 ℃(130 ℃~131 ℃[5]);Rf=0.52[展开剂:V(乙醇) ∶V(二氯甲烷) ∶V(氨水)=3.0 ∶1.0 ∶0.1,下同];1H NMRδ: 1.59~1.43(m, 3H), 1.72~1.88(m, 1H), 1.95~1.99(t, 2H), 2.44~2.21(m, 2H), 2.56(s, 3H, CH3), 2.74~2.79(q, 1H, CH), 2.99~3.09(m, 3H), 9.42(s, 1H, =N OH);13C NMRδ: 8.00(9C), 7.65~7.77(1C, 5C), 2.00(7C), 1.08~1.15(2C, 4C, 6C, 8C), 12.57(3C)。
(4)endo-1的合成
在四口烧瓶中加入LiAlH43.4 g的无水THF(55 mL)溶液,搅拌下于-10 ℃~-5 ℃滴加浓硫酸3.0 mL的THF(11.25 mL)溶液,反应2 h;于30 ℃滴加54.5 g(27 mmol)的THF(80 mL)溶液(约30 min),滴毕,于40 ℃反应8 h。冷却至-10 ℃,小心加入H2O(15 mL)和THF(15 mL)的混合溶液,于30 ℃反应1.5 h。过滤,滤饼用二氯甲烷洗涤,合并滤液与洗液,用无水硫酸镁干燥过夜,蒸除溶剂得黄色液体endo,exo-14.0 g,还原收率96.6%(65.4%[5], 93%[7], 92%[8])。
endo,exo-1经柱层析[洗脱剂:V(乙醇) ∶V(二氯甲烷) ∶V(氨水)=3.0 ∶1.0 ∶0.1]]分离得淡黄色液体endo-13.05 g(占endo,exo-1的76.06%),收率73.5%, b.p.200 ℃/101 kPa,Rf=0.21;1H NMRδ: 3.25~3.29(m, 1H, CHNH2), 3.00~3.03(m, 2H, NCH), 2.45(s, 3H, CH3), 2.26~2.36(q, 2H, CH2), 1.46~1.96(m, 4H, CH2), 1.25~1.99(m, 4H, CH2);13C NMRδ: 13.9(1C), 23.8(2C), 36.5(2C), 39.9(1C), 42.1(1C), 51.2(2C); ESI-MSm/z: 155{[M+H]+}。
2 结果与讨论
在3的合成中,文献方法采用发烟硫酸[12],或20%的发烟硫酸[5],由于发烟硫酸存在环境污染较大,操作不便等缺点,因此改用浓硫酸[6],由于反应不断有水生成,会降低硫酸活性,影响产率。鉴于以上两点,本文作了改进,采用混酸[V(98% H2SO4) ∶V(20%发烟H2SO4)=3∶1],一方面有效降低了SO3在硫酸中的含量,不会有大量的硫酸雾,操作较方便;另一方面在浓硫酸中加入一定浓度的发烟硫酸后,由于SO3能吸收反应生成的水,有利于该反应进行。虽然产率较文献值[12]低,但具有更好的经济性和可操作性。
合成4时,由于反应历程比较复杂,副产物较多,延长反应时间对提高收率有利,实验结果表明反应时间以48 h为宜。并且本文采用简单的重结晶代替文献[5]方法的减压蒸馏提纯方法,操作更简单。收率略有提高。
采用文献[7]方法合成5,由于直接将盐酸羟胺固体加入反应瓶中,固体在有机相中不容易溶解,致使产率低。本文则通过盐酸羟胺与醋酸钠的反应生成醋酸和羟胺,再进行后面的反应,能够大大提高收率。实验结果表明多加醋酸钠,产率最高能达到73%。为得到纯度更高的产品,可采用简单的乙酸乙酯重结晶方法提纯。
使用变性LiAlH4作为氢化还原试剂[9]合成1,由于变性LiAlH4体积较大,进攻外侧的空间位阻比进攻内侧小,所以得到的大多数产物都是exo-1。实验结果表明,用摩尔数3倍于原料的变性LiAlH4还原,并适当延长反应时间,endo-1产率能达到96.6%,且纯度较高。

[1] 徐积恩. 止吐药格雷西隆(Granisetron)[J].国外医药—合成药、生化药、制剂分册,1992,13(5):310-311.
[2] 李家明,周思祥,章兴. 盐酸格拉司琼的合成[J].中国医药工业杂志,2000,31(2):49-50.
[3] 时颖华,王宏图. 格拉司琼的临床研究进展[J].上海医药,1997,(12):21-24.
[4] 周斌. 5-HT3受体拮抗剂止吐药的研究开发概况[J].药学进展, 1992, 16(4): 210.
[5] 张毅. 一种制备格拉司琼及其盐的方法[P].CN 1 190 436C,2003.
[6] 周贤言,潘联根. 一种丙酮二羧酸酯的制备方法[P].CN 101 475 482,2009.
[7] SMITH G E. Process for the production of aminozaobicycloalkanes from oximes[P].WO 9 603 401,1994.
[8] Benzoxazole carboxamides for treating CINV and IBS-D[P].US 2 006 183 769,2006.
[9] Peter D, Gunter E, Bruno H,etal. Bridge piperridyl esters and amides[P].GB 2 125 398,1983.
[10] AMETA S C. Dye-sensitized photo oxygenation of citric acid by singlet oxygen[J].Oriental Journal of Chemistry,1989,5(2):164-167.
[11] Cope A C, Overberger C G. Synthesis of cyclooctatetraene from pseudopelletierine[J].Journal of the American Chemical Society,1948,70:1433-1437.
[12] Roger Adams,H. M.Chiles,etal. Acetonedicarboxylic acid[J].Organic Syntheses,1925:5.
猜你喜欢
北京大学学报(自然科学版)(2022年3期)2022-06-17
中国环境科学(2021年8期)2021-09-03
化工生产与技术(2021年6期)2021-02-18
上海化工(2018年10期)2018-10-31
化学反应工程与工艺(2017年6期)2017-06-12
医药导报(2015年9期)2015-02-11
中国继续医学教育(2015年4期)2015-01-31
化工生产与技术(2014年5期)2014-02-27
西南军医(2014年4期)2014-01-19
中国产前诊断杂志(电子版)(2013年1期)2013-01-21
