
吉非替尼的合成工艺研究
2011-11-21 08:25张道旭周伟新
中国现代药物应用 2011年3期
张道旭 周伟新
吉非替尼的合成工艺研究
张道旭 周伟新
目的 优化吉非替尼合成工艺。方法 以3-羟基-4-甲氧基苯甲酸为起始原料,经过酯化、缩合、硝化、还原、环合、氯代、氨化、取代等步骤合成吉非替尼。结果 反应总收率约为42.8%,目标产物经过红外光谱、核磁共振氢谱、质谱、元素分析确证结构。结论 本工艺反应条件温和,操作简便,收率高,适用于工业化生产。
吉非替尼;表皮生长因子受体;药物化学;工艺改进
吉非替尼--选择性表皮生长因子受体(EGFR)酪氨酸激酶抑制剂,由阿斯利康公司首先研制,商品名为易瑞沙(IressaTM),化学名为 4-(3-氯-4-氟苯胺基)-7-甲氧基-6-(3-吗啉基丙氧基)喹唑啉。其通过阻断酪氨酸激酶信号传导通路抑制肿瘤的生长,从而发挥抗癌作用。主要用于非小细胞肺癌的治疗[1],于2003年经FDA批准在美国上市,具有广阔的市场前景。
根据文献报道[4],以3-羟基-4-甲氧基苯甲酸甲酯为起始原料,经过缩合、硝化、还原、环合、氯代、取代等7步反应制备得到目标产物1,此合成路线副反应少,产品质量易于控制,但用混酸进行硝化时易发生爆炸等危险,利用铁粉还原硝基时会产生大量铁泥,即不易过滤又增加了三废处理的难度,不利于工业化生产。因此,参考文献[2,3,5]重点对硝化和还原两步进行了工艺改进,使新的工艺路线具有反应条件温和、操作简便易行、收率高、纯度好,环境污染小等优点,更加适合工业化生产。具体合成路线见图1。
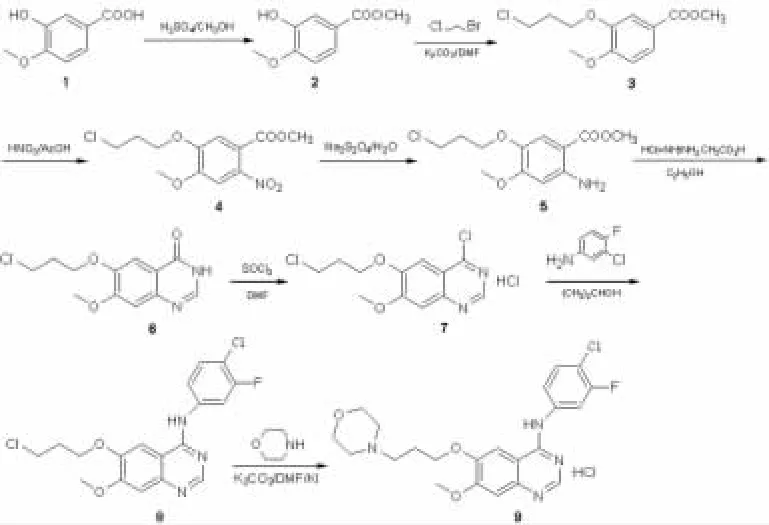
图1 The synthetic rount of target compound
1 实验部分
1.1 仪器与试剂
WRS-1A型数字熔点仪;IFS-55(Bruker公司)红外光谱仪;ACF-300(Bruker公司)核磁共振仪(TMS为内标);Agilent LC/MSD型液质联用(LS-MS);Carlo Erba-1106型自动元素分析仪。
所采用试剂均为化学纯。
1.2 合成
1.2.1 3-羟基-4-甲氧基苯甲酸甲酯(2)的合成 将16.7 g(0.1 mol)3-羟基-4-甲氧基苯甲酸(1),2 ml 98%浓硫酸加入150 ml无水甲醇中,回流反应8 h。蒸去溶剂,得到棕黄色油状物,再加入100 ml氯仿溶解,5%碳酸氢钠洗至pH值至中性,水洗,无水硫酸钠干燥,蒸去溶剂得到淡黄色产物17.8 g,收率98%,m.p.64℃ ~67℃。
[1]Levin M,DcSouza N,Castaner J.Iressa oncolytic EGF receptor tyrosine kinase inhibitor.Drugs Fut,2002,27(4):339-345.
[2]Kleth HG.Quinazoline derivatives[P]:US 5 770 599,1996.
[3]BRIDGES AJ,ZHOU HR.Tyrosie kinase inh ibito rs.8.An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxy quinazo-line(PD153035),a potent inhibitor of the epidermal growth factor receptor.J.Med.Chem,1996,39(1):267-276.
[4]袁立,郝金恒,张勇,等.4-(3-氯-4-氟苯胺基)-7-甲氧基-6-(3-吗啉基丙氧基)喹唑啉的合成.中国药物化学杂志,2005,15(1):39-41.
[5]谢良辉,欧阳贵平.吉非替尼的合成工艺改进.合成化学,201018(4):523-525.
150056 哈药集团制药六厂(张道旭);鹤矿集团总医院(周伟新)
1.2.2 4-甲氧基-3-(3-氯丙氧基)苯甲酸甲酯(3)的合成
将 9.1 g(0.05 mol)化合物(2),11.85 g(0.075 mol)1,3-溴氯丙烷和10.35 g(0.075 mol)无水碳酸钾加入100 ml DMF中,于40℃反应4 h。将反应液加入500Ml水中,再用二氯甲烷40 ml分两次进行萃取,合并有机相、水洗、无水硫酸钠干燥,蒸去溶剂,得灰色产物 12.5 g,收率 96.6%,m.p.51℃~54℃。
1.2.3 2-硝基-4-甲氧基-5-(3-氯丙氧基)苯甲酸甲酯(4)的合成 将48 ml 70%浓硝酸(0.72 mol)加入500 ml反应瓶中,升温至45℃ ~50℃,搅拌下缓慢加入12.5 g化合物(3)的醋酸(200 ml)溶液,反应8 h。倒入冰水中,用5%碳酸氢钠水溶液调pH值至7.5~8,过滤、滤饼水洗、干燥,得到黄色产物13.6 g,收率92.8%,m.p.68℃ ~71℃。
1.2.4 2-氨基-4-甲氧基-5-(3-氯丙氧基)苯甲酸甲酯(5)的合成 将13.6 g(0.045 mol)化合物(4)溶于300 ml水中,搅拌下加入26.6 g(0.15 mol)硫代硫酸钠,升温至50℃反应3 h后再升温至70℃,3 h内缓慢滴加68 ml 25%盐酸。反应毕,降至0℃,用50%氢氧化钠水溶液调pH值至10,再倒入100 ml二氯甲烷中,分相,有机相水洗至中性,无水硫酸钠干燥,蒸去溶剂,得到灰白色产物 11.7 g,收率95.1%,m.p.82℃ ~85℃。
1.2.5 7-甲氧基-6-(3-氯丙氧基)喹唑啉-4(3H)-酮(6)的合成 将14.2 g(0.05 mol)化合物(5)和8.1 g(0.075 mol)醋酸甲脒加入150 ml无水乙醇中,回流反应16 h。反应液降至室温,过滤,滤饼用少量无水乙醇洗,干燥,得白色产物12.8 g,收率92.0%,m.p.212℃ ~214℃。
1.2.6 4-氯-7-甲氧基-6-(3-氯丙氧基)喹唑啉(7)的合成
将13.4 g(0.05 mol)化合物(6)和4 ml DMP加入到150 ml二氯亚砜中,升温至回流反应4 h。降至室温,蒸去溶剂,得到黄色油状物,搅拌下加入200 ml冰水中,过滤,水洗滤饼至中性,干燥,得黄色产物 12.6 g,收率 88.1%,m.p.205℃~208℃。
1.2.7 4-(3-氯-4-氟苯胺基)-7-甲氧基-6-(3-氯丙氧基)喹唑啉(8)的合成-将14.4 g(0.05 mol)化合物(7)和8.8 g(0.06 mol)3-氯-4-氟苯胺加入到250 ml异丙醇中,升温至45~50℃反应4 h。降至室温,过滤,滤饼用少量异丙醇洗,干燥,得浅黄色产物 17.0 g,收率85.7%,m.p.225 ~228℃。
1.2.8 4-(3-氯-4-氟苯胺基)-7-甲氧基-6-[3-(4-吗啉基) 丙氧基]喹唑啉(9)的合成 将10 g(0.025 mol)化合物(8),4.2 g(0.03 mol)无水碳酸钾和3.0 g碘化钾加入到100 ml DMF中,搅拌0.5 h,再滴加2.6 g(0.03 mol)吗啉的DMF混合溶液(25 ml),升温至75℃反应5 h。将反应液倒入冰水中,用二氯甲烷(50 ml*2)萃取,合并有机相,水洗至中性,无水硫酸钠干燥,蒸去溶剂,得淡黄色产物9.0 g,收率73.7%,m.p.118℃ ~119℃(文献[2]m.p.119 ~ 120℃)。IR(KBr)υ(cm-1):3445,3015,1625,1450,1475,950,825,770;1H-NMR(DMSO-d6)δ 9.86(1H,s,N-H),8.64(1H,d,Ar-H),8.25(1H,m,Ar-H),7.90(1H,m,Ar-H),7.82(1H,s,Ar-H),7.26(1H,t,Ar-H),7.11(1H,s,Ar-H),4.21(2H,t),4.01(3H,s,OCH3),3.61(4H,t),2.48(2H,m),2.47(4H,s),1.96(2H,m);MS m/z 447.2[M+1](C22H24ClFN4O3)理论值(%):C 59.17,H 5.40,N 12.55; 测定值(%):C 60.23,H 5.34,N 12.60。
猜你喜欢
电子乐园·上旬刊(2022年5期)2022-04-09
农药科学与管理(2019年8期)2019-11-23
天然产物研究与开发(2018年8期)2018-09-10
铜仁学院学报(2018年6期)2018-07-05
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
中国药业(2014年17期)2014-05-26

- 中国现代药物应用的其它文章
- 拉西地平精制工艺的改进
- 碱式水杨酸铋干混悬剂的溶出度测定
- 浅谈数学模型在医学领域的应用