
钛表面含硅羟基磷灰石涂层的电化学合成和表征
2011-11-09 10:42:48李登虎林东洋王小祥
无机化学学报 2011年6期
李登虎 林 军 林东洋 王小祥*,
(1浙江大学材料科学与工程学系,杭州 310027)
(2浙江大学医学院附属第一医院口腔科,杭州 310003)
钛表面含硅羟基磷灰石涂层的电化学合成和表征
李登虎1林 军2林东洋1王小祥*,1
(1浙江大学材料科学与工程学系,杭州 310027)
(2浙江大学医学院附属第一医院口腔科,杭州 310003)
在含有Ca2+,P以及Si的电解液中,通过电化学恒电位方法,在工作电压为3 V温度为85℃的条件下沉积1 h,于钛表面上制得含硅羟基磷灰石涂层。通过电感耦合等离子体原子发射光谱(ICP)、扫描电镜(SEM)、X-射线衍射(XRD)、探针式轮廓仪(SP)、红外光谱(FTIR)对涂层进行分析。结果表明:电化学恒电位方法可制得Si饱和含量为0.55wt%左右的Si-HA涂层,Si以Si形式取代P进入HA晶格,造成羟基磷灰石中OH-减小以维持电荷平衡。另外,电解液中Si元素的存在抑制涂层中HA晶体的生长,使涂层变薄,且当电解液中nSi/(nSi+nP)达到20%时Si-HA晶体形貌由单独的棒状转变为根部相连的树枝状。
羟基磷灰石;硅掺杂;电化学沉积;涂层
羟基磷灰石(hydroxyapatite,HA)是人体和动物骨骼、牙齿的主要无机成分,具有良好的生物相容性和化学稳定性,植入人体后,能与人体骨骼组织形成化学键合,因此被广泛应用于钛和钛合金表面涂层,以增加钛和钛合金的表面生物活性[1-2]。但相对于生物玻璃或A-W玻璃陶瓷等生物活性材料,纯HA与骨组织之间的反应性较低,前期与骨发生整合的速率也相对较低,致使病人治愈时间加长[3]。另外,作为磷酸钙中最稳定的相,纯HA在体内的吸收度低,长时间的遗留会造成种植体在体内的固定缺陷[4]。在实际人骨中,其矿物相往往是含有碳酸根、钠、镁、硅、锌、锶等掺杂离子的HA。为了改善纯HA的生物活性,其中一种方法就是得到成分与实际骨成分相同的HA,即向HA中掺杂这些离子。而在这些掺杂元素中,硅普遍被认为是人体必需元素,Carlise的研究表明硅对早期的骨矿化过程和软组织的形成起着非常重要的作用[5]。体内和体外实验研究已经证明在HA中掺杂硅元素能够有效的提高成骨细胞的活性并且骨的生长速率要比纯HA高14.5%[6-7]。
现有的制备含硅羟基磷灰石(Ca10(PO4)6-x(SiO4)x(OH)2-x,Si-HA)涂层方法中,仿生法周期过长[8],等离子体喷涂不能对复杂形状的种植体进行作业[9],激光熔覆法容易出现局部区域温度过高导致涂层分解[10]。近年来,在HA涂层的制备方法中,电化学方法由于其成本低廉、反应条件温和、沉积效率高、非线性工艺、成分可控等优点[11],受到了学者们广泛的重视。Jiao等[12]已成功的在钛基体上采用电化学方法制备出含镁HA涂层,表明了电化学方法是一种有效的制备掺杂离子HA涂层的方法。目前此方法制备掺杂离子HA涂层还未被广泛研究,特别是制备掺杂阴离子(硅酸根)HA涂层。本文利用电化学恒电位方法,以 CaCl2,、NH4H2PO4、NaCl和 Na2SiO3· 9H2O为原料,在钛表面制备含硅HA涂层,系统的研究了该方法中电解液所含硅含量对涂层结构的影响,并对原因进行了初步探讨。
1 实验方法
1.1 电化学合成Si-HA涂层
尺寸为10 mm×10 mm×1 mm商业用的纯钛经400#到800#砂纸打磨,经丙酮清洗后,用体积比VHF∶VHNO3∶VH2O=1∶3∶10酸液酸蚀1 min,最后用去离子水清洗,并接在阴极上,阳极采用铂电极。电解液用分析纯 CaCl2、NH4H2PO4、NaCl和 Na2SiO3·9H2O配制。其中Ca2+浓度固定为1.2 mmol·L-1,沉积过程中假定Si元素取代HA中的P元素,固定 (P+ S)浓度为0.72 mmol·L-1,并调整Si/(P+ Si)物质的量比nSi/(nSi+nP)分别为0%、1%、2%、3%、4%、5%、10%、20%、30%,Cl-浓度为0.1 mol·L-1用以增加溶液的导电性。电化学工作站(CHI1140A)工作电压恒定设定为3.0 V,电极距离控制在2 cm,用盐酸氨水调节所有电解液pH=6,并将温度都设为85℃,沉积时间均为1 h。
1.2 Si-HA涂层的表征
涂层元素含量用XPS型等离子电感耦合原子发射光谱(ICP-AES,美国热电公司)测定,测试时将涂层溶解到25 mL 0.1 mol·L-1的HCl中 (每个ICP测试样品溶解5片涂层)。用Philips XD-98 X射线衍射仪(XRD,Cu靶Kα线,电压40 kV,管流40 mA,步长0.020,扫描速度4°·min-1)检测物相。涂层厚度用Dektak 3探针式轮廓仪检测。涂层用刀片刮下,加入KBr压片处理,用AVATAR傅里叶变换红外光谱仪(FTIR)表征化学基团。最后用Hitachi S4800型场发射扫描电镜(SEM)观测涂层表面形貌。
2 结果与讨论
2.1 沉积涂层元素含量分析
实验测定Ca、P、Si 3个元素的含量,以Ca2+离子浓度为标准,涂层总量假设沉积涂层全部为纯HA计算,最终涂层中的Si含量数据如图1所示。由图可见,当电解液中nSi/(nSi+nP)低于3%时,所得涂层中Si含量随电解液中Si含量的增加呈直线上升趋势。而当电解液中nSi/(nSi+nP)超过3%后,所得涂层中的Si含量增加开始变缓,并逐渐趋向饱和状态,在电解液的nSi/(nSi+nP)为10%的时候达到饱和值(大约0.55wt%)。而在10%以后,继续增加电解液中的Si含量,涂层中的Si含量维持不变。此结果表明电化学恒电位方法可制备Si-HA涂层,且涂层中Si含量可控。涂层中除Si含量以外,nSi/(nSi+nP)比也非常重要。该比值在电解液中nSi/(nSi+nP)小于20%时,与涂层中Si的含量表现出相同的趋势,但在20%以后,却呈现上升趋势,可能与涂层中Si-HA晶体结晶度下降有关。

图1 涂层中的Si成分分析Fig.1 Measured Si wt%and nSi/(nSi+nP)ratio in coatings
与等离子喷涂[9]所得Si-HA涂层的Si掺杂水平(大于1wt%)相比,本方法所制得的涂层Si含量偏低,说明电化学恒电位方法在本实验条件中较难引入大量Si元素到HA涂层。其原因可能是:(1)Si离子半径比P离子半径大很多,Si离子进入到HA中P离子的位置需要很高的能量,其他高Si含量的Si-HA制备方法中都会涉及到高温处理工艺,以此来提高HA中的Si含量,而本方法中85℃的温度条件较难满足此需求;(2)由于硅酸根离子带负电,阴极附近会排斥硅酸根离子,导致钛基体附近硅酸根离子的浓度过低,从而影响最终涂层的Si含量。然而,HA中高Si含量并不意味着高的生物性能。Carlise已经证明,在早期的骨钙化过程中,检测到Si的最高含量为0.5wt%[5]。Vallet-Regi等[13]指出0.5wt%的Si含量已足够产生非常重要的生物改性。Hing等通过为期12周的小模型研究,证明了Si含量为0.8wt%时,骨的形成是最佳的[14]。此外若材料中含有太多的Si,会在局部区域产生过高的Si浓度,对周围的组织产生毒害[15]。
2.2 沉积涂层物性分析
图2是涂层的XRD图。图中XRD的主要峰值(除钛基体外)与HA的PDF卡片No.9-432的峰吻合,电解液中Si元素的添加并没有使涂层出现如Ca2SiO3的杂相,涂层主要晶体相为HA。当电解液中Si含量增加时,涂层中HA主要峰型的相对强度逐渐减弱,特别是HA的(002)峰。原因可能有:(1)涂层的Si-HA结晶度下降,晶体特征减少,出现非晶;(2)Si-HA涂层的总含量减小,涂层变薄,使得衍射峰强度减小。此外,较强(002)晶面峰表明了HA晶体是沿c轴方向垂直于钛基体表面择优生长。
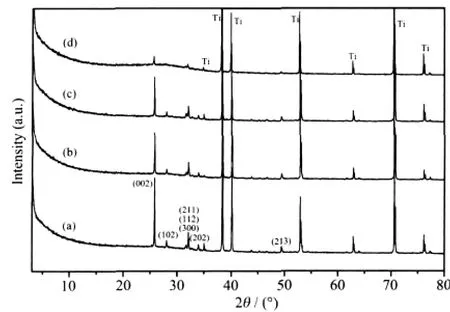
图2 涂层XRD图Fig.2 XRD patterns of the coatings deposited on titanium in the electrolytes with various nSi/(nSi+nP)molar ratio:(a)0%,(b)5%,(c)10%,(d)20%
涂层的厚度变化如图3所示,从图中可以看出,Si元素的添加使得涂层厚度逐渐变薄,纯HA的厚度在3.7 μm左右,再添加5%和10%nSi/(nSi+nP)的Si后,涂层厚度分别减少到2 μm和1.4 μm,而在含20%nSi/(nSi+nP)的Si的电解液中制得的Si-HA涂层厚度低至0.7 μm。在大部分的Si-HA制备方法中,Si元素的存在会抑制HA晶体的生长,使Si-HA晶粒尺寸减小。涂层厚度变化表明在电化学沉积中,HA择优生长方向(002)方向生长(即沿涂层厚度方向)抑制作用要比其他方向强,Si含量越多,抑制作用越明显,涂层越薄。此抑制作用被认为是引起XRD图中HA峰型相对强度降低的主要原因,且亦有可能是导致涂层中nSi/(nSi+nP)比上升的原因之一。

图3 涂层的厚度变化Fig.3 Change of coatings thickness
涂层的FTIR图谱如图4所示。HA典型的吸收峰在各个涂层中都能被观测到,630、3 570 cm-1为OH-的特征峰,507、603、1 089、1 038和962 cm-1为P的特征峰。另外在1 456,1 410和875 cm-1为 C的特征峰,表明所制得的HA涂层为含碳酸根的HA。

图4 涂层的FTIR图谱Fig.4 FTIR spectra of deposits scraped from the titanium surface under different preparation conditions with various nSi/(nSi+nP)molar ratio:(a)0%,(b)5%, (c)10%,(d)20%
对比纯HA和Si-HA涂层的FTIR图谱,结果显示Si元素的添加修正了800~1100 cm-1和500~700 cm-1的P特征峰,并随着Si含量的增加,962 cm-1和470 cm-1的P峰强度减小。OH-在3570 cm-1和630 cm-1的峰也随着Si含量的增加逐渐的减小。这与Gibson等[16]提出的Si掺杂HA电荷平衡理论模型非常吻合。在该电荷平衡理论模型中,Si首先以S形式存在,取代部分P,然后与P一起生成Si-HA,由于电荷平衡的需要,会丢失部分的OH-,造成Si-HA与纯HA相比P和OH-相应的减少。由于S与P价态差异,S取代P是耦合的[17],这也是涂层中Si元素含量能有饱和值并且偏低的一个重要原因。
在FTIR图谱中未发现Si-O键的存在,可能是其峰位与强HA峰重合无法分峰,尤其是在Si-O键振动强烈的区域1100和1085~1000 cm-1之间[18],还有可能是Si含量太少导致Si-O键峰难以辨认。图谱中出现的碳酸根峰,可能是由涂层制备过程中电解液中吸收的CO2参与电沉积反应所得[19]。涂层中碳酸根的存在可能会阻止硅酸根进入HA晶格,因为碳酸根比硅酸根更容易进入HA的晶格[17,20],碳酸根的出现可能也是导致涂层中Si含量偏低的一个原因。
2.3 涂层形貌分析

图5 涂层的表面微观形貌Fig.5 SEM images of the coatings prepared under different nSi/(nSi+nP)molar ratio:(a)0%,(b)5%,(c)10%,(d)20%

图6 树枝状晶体的早期生长过程SEM图Fig.6 SEM images of the coatings of dendritic crystals at primary stage:(a)8 min,(b)10 min,(c)12 min
涂层的微观形貌见图5,涂层由规则的六棱柱棒状晶体组成,六棱柱晶体直径大约为100 nm,是典型的HA晶体形貌。当电解液中加入在少量Si时,晶体仍保持单独的六棱柱形貌,晶体尺寸没有变化,只引起晶体排列更加紧密。而当电解液中Si元素含量达到20%时,涂层中的HA晶体形貌开始由单独的六棱柱棒状晶体转变为根部连接在一起的树枝状晶体,但树枝状晶体的前端六棱柱部分尺寸仍维持不变,如图4(d)所示。树枝状晶体出现的原因可能与Si元素对涂层HA晶体生长起抑制作用有关,其前期生长过程如图6所示。从图中可以看出,HA并不是直接在钛基体上形核然后长大,而是先以片状磷酸八钙(OCP)前躯体的形式存在,该前驱体已在很多文献中报道过[21]。在持续的电化学过程中OCP再转变为HA,并且单片OCP可转变为多个HA。单片片状OCP晶体转变为多个纳米棒状HA晶体后,由于这些棒状晶体处在纳米级别容易团聚,加上Si元素的抑制作用,HA晶体纵向生长缓慢,导致沉积完后的棒状HA晶体未完全分离,但在长时间的沉积过程中完成横向生长,进而出现前端六棱柱尺寸未变的树枝状晶体。该结果与XRD图中(002)峰相对强度逐渐降低结果一致。
2.4 沉积过程分析
根据上述分析,电化学恒电位沉积过程中,钛基体表面发生的反应如下[8,22]:

反应开始先电解水产生OH-,由于OH-得存在将会产生P。同时,Si与H2O反应,最终以Si形式存在在电解液中。最后在钛基体上形成Ca10(PO4)6-x(SiO4)x(OH)2-x化合物,也就是Si-HA涂层。
3 结 论
通过电化学恒电位方法,以CaCl2,、NH4H2PO4、NaCl和Na2SiO3·9H2O为原料,在钛基体上成功合成Si-HA涂层:涂层中Si元素含量偏低,在电解液中nSi/n(Si+P)达到10%时,涂层Si含量达到饱和值0.55wt%左右,继续增加电解液中Si含量,涂层中Si含量维持不变,但涂层中的nSi/(nSi+nP)会进一步增加;电解液中Si元素以Si形式存在,取代P,进入HA的晶格,HA丢失部分OH-以维持电荷平衡;电解液中Si元素的存在会抑制涂层中HA晶体的生长,特别是垂直钛基体的方向,导致涂层变薄,且由于该抑制作用,当电解液中的nSi/(nSi+nP)为20%时,涂层中Si-HA晶体的形貌从单独的棒状转变为根部相互连接的树枝状。
[1]Ye W,Wang X X.Mater.Lett.,2007,61:4060-4065
[2]Ma M H,Ye W,Wang X X.Mater.Lett.,2008,62:3875-3877
[3]Pietak A M,Reid J W,Stott M J,et al.Biomaterials,2007, 28:4023-4032
[4]Mastrogiacoma M,Muraglia A,Komlev V,et al.Orthod. Craniofacial.Res.,2005,8:277-284
[5]Carlisle E M.Science,1970,167:279-280
[6]Patel N,Best S,Bonfield W,et al.J.Mater.Sci.:Mater. Med.,2002,13:1199-1206
[7]Gibson I,Hing J A,Best S,et al.Bioceramics,1999,12:191-194
[8]Zhang E,Zou C,Zeng S.Surf.Coat.Technol.,2009,203:1075 -1080
[9]Tang Q,Brooks R,Rushtion N,et al.J.Mater.Sci.:Mater. Med.,2010,21:173-181
[10]Yang Y L,Paital S R,Dahotre N B.J.Mater.Sci.:Mater. Med.,2010,21:2511-2521
[11]YANG Cheng-Xin(杨成鑫),LIN Dong-Yang(林东洋),JIANG Yong(江勇),et al.J.Inorg.Mater.(Wuji Cailiao Xuebao), 2010,5(2):206-210
[12]Jiao M J,Wang X X.Mater.Lett.,2009,63:2286-2289
[13]Vallet-Regí M,Arcos D.J.Mater.Chem.,2005,15:1509-1516
[14]Hing K,Revell P,Smith N,et al.Biomaterials,2006,27: 5014-5026
[15]Nagase M,Abe Y,Chigira M,et al.Biomaterials,1992,13: 172-175
[16]Gibson I,Best S,Bonefield W.J.Biomed.Mater.Res.A, 1999,44:422-428
[17]Tang X L,Xiao X F,Liu R F.Mater.Lett.,2005,59:3841-3846
[18]Andersson J,Areva S,Spliethoff B,et al.Biomaterials,2005, 26:6827-6835
[19]Rößler S,Sewing A,Stölzel M,et al.J.Biomed.Mater.Res. A,2002,64:653-663
[20]TANG Xiao-Lian(唐晓恋),XIAO-Xiu-Feng(肖秀峰),LIU Rong-Fang(刘榕芳).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2005,21(10):1500-1504
[21]Xin R L,Leng Y,Wang N.J.Cryst.Growth,2006,289:339-344
[22]Lin D Y,Wang X X.Surf.Coat.Tech.,2010,204:3205-3213
Synthesis and Charaterization of Silicon-Substituted Hydroxyapatite Coating by Electrochemical Deposition on Ti Substrate
LI Deng-Hu1LIN Jun2LIN Dong-Yang1WANG Xiao-Xiang*,1
(1Department of Material Scienece and Engineering,Zhejiang Unviersity,Hangzhou310027,China)
(2Department of Stomatology,The First Affiliated Hospital,College of Medicine,Zhejiang Unviersity,Hangzhou310003,China)
Silicon-substituted hydroxyapaptite coatings were prepared on titanium substrate with electrochemical deposition technique in electrolytes containing Ca2+,Pand Siions.The deposition was all conducted at a constant voltage of 3.0 V for 1 h at 85℃.The as-prepared coatings were examined by inductively coupled plasma (ICP),X-ray diffraction(XRD),stylus profiler(SP),Fourier transform infrared spectroscopy(FTIR),field-emission-type scanning electron microscope (SEM).The results show that the silicon amount in Si-HA coatings deposited by electrochemical method can reach to a saturation level of around 0.55wt%,and the substitution of silicate for phosphate in the form of Sresults in loss of some OH-to maintain the charge balance.Additionally,the presence of silicon ions in electrolytes inhibits HA crystals growth in coatings,leading to decrease of coating thickness and transformation of rod-like crystals into dendritic crystals in the electrolyte of 20%molar ratio silicon.
hydroxyapatite;silicon-substitution;electrodeposition;coating
TB333;O646
A
1001-4861(2011)06-1027-06
2010-12-09。收修改稿日期:2011-01-25。
国家自然科学基金(No.50571088)资助项目。
*通讯联系人。E-mail:msewangxx@zju.edu.cn
猜你喜欢
人工晶体学报(2021年10期)2021-11-26 02:39:18
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19 09:03:04
山东冶金(2019年5期)2019-11-16 09:09:12
表面工程与再制造(2019年6期)2019-08-24 06:40:08
资源节约与环保(2018年1期)2018-02-08 02:18:27
池州学院学报(2017年3期)2017-10-16 01:38:37
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
机械制造与自动化(2014年1期)2014-03-01 04:21:57
少儿科学周刊·儿童版(2013年2期)2013-05-13 09:21:06
