
Mg和Zn掺杂CuAlS2电子结构的分析*
2011-11-02 03:25万文坚姚若河耿魁伟
物理学报 2011年6期
万文坚姚若河耿魁伟
Mg和Zn掺杂CuAlS2电子结构的分析*
万文坚 姚若河耿魁伟
(华南理工大学电子与信息学院,广州510640)
(2010年5月29日收到;2010年10月8日收到修改稿)
从能带结构和态密度分析了黄铜矿CuAlS2的电子结构.对比未掺杂CuAlS2,从晶体结构、电子结构、电荷密度分布讨论了Mg和Zn替位Al掺杂对CuAlS2的影响.结果表明:Mg和Zn掺杂CuAlS2都导致晶格常数增大,Mg掺杂晶胞体积增大更多;掺杂在价带顶引入受主态,形成p型电导;Mg掺杂比Zn掺杂的受主能级电离能略小;而Zn掺杂CuAlS2体系总能更低,晶格结构更稳定.
CuAlS2,p型掺杂,电子结构,能带结构
PACS:71.20.Nr,71.20.-b,61.72.U-,68.55.Ln
1.引言
MⅠ-MⅢ-MⅥ2(MⅠ=Cu,Ag;MⅢ=Al,Ga,In; MⅥ=S,Se,Te)黄铜矿系化合物[1—3]为直接带隙半导体,是一类优良的光电子材料.其中Cu AlS[4—6]2的室温禁带宽度为3.49 eV,激子束缚能为70 meV,比ZnO,Zn S以及GaN的激子束缚能更大,可用于高效的紫外光发射;CuAlS2由于价带顶产生Cu 3 d与S 3 p轨道杂化,S对空穴的局域化作用被削弱,空穴可较自由地移动,具有比CuAlO2更好的p型导电性能[7].
关于Cu AlS2的掺杂改性已有一些研究.贫铜CuAlS2材料[7]尽管能够产生更多铜空位,但由于破坏了Cu—S网络载流子输运路径,p型导电性能反而更差;富硫CuAlS2材料[8]相比于化学计量配比体相,载流子浓度增加,p型导电性增强,体材料电导率达到4.6 S·cm-1.用Mg替位Al,放电等离子烧结制备掺杂CuAlS2体材料[9],电导率可达41.7 S· cm-1;用Zn替位Al,渠道火花烧蚀制备掺杂CuAlS2薄膜[10],电导率高达63.5 S·cm-1;脉冲等离子沉积制备Zn掺杂薄膜[11],薄膜电导率达到50.9 S· cm-1.上述实验表明,Mg以及Zn能够显著增强CuAlS2导电性能,但目前尚未有Mg或Zn掺杂CuAlS2理论研究的详细报道.本文采用第一性原理平面波赝势法(PWP)[12]对未掺杂及Mg和Zn掺杂Cu AlS2的能带结构、态密度分布和差分电荷密度进行了计算和分析.
2 .理论模型和计算方法
稳定相CuAlS2为四方晶系黄铜矿结构,属于I42 d空间群.晶格常数a=b=5.3336,c= 10.4440,c/a=1.958[13].Cu AlS2惯用晶胞如图1所示,包含有16个原子,每个原子均为4配位:1个S与2个Al和2个Cu配位,1个Cu或1个Al与4个S配位.本文计算考虑Al的替位掺杂,采用2×2 ×1超原胞,共包含64个原子,用1个Mg原子或1个Zn原子取代超原胞中的1个Al原子进行掺杂计算,掺杂率为6.25%.
计算由基于密度泛函理论的从头算量子力学程序CASTEP[14]来完成.采用超软赝势描述离子实与价电子之间的相互作用,选取各元素的价电子组态分别为Cu:3d104 s1,Al:3s23 p1,S:3 s23 p4,Mg:2 p63s2,Zn:3d104 s2.平面波截止能Ecut选取为300 eV,交换-关联能采用改进的广义梯度近似(PBE-GGA)来处理.总能量和电荷密度在布里渊区的积分使用Monkhorst-Pack方案,选择k网格为2×2×2,快速傅里叶变换(FFT)网格取54×54×54.自洽场运算采用Pulay密度混合法,自洽场精度设为每原子2×10-6eV.采用BFGS算法对模型的几何结构进行优化,优化参数如下:原子间相互作用力收敛标准为0.05 eV/,单原子能量收敛标准为2×10-5eV,原子最大位移收敛标准为0.002,晶体内应力收敛标准为0.1 GPa.

图1 CuAlS2晶胞示意图
3.CuAlS2的电子结构
图2和图3分别给出了CuAlS2的能带结构和态密度分布,包括各元素的分波态密度和总体态密度.从图2可以看出黄铜矿CuAlS2的价带由-7.1 eV到-3.3 eV的下价带和-1.9 eV到0 eV的上价带组成,价带顶出现3个子带,最上面的子带与下面2个子带由晶体场分裂所致,下面2个子带由自旋-轨道耦合分裂形成[15],3个子带在布里渊区中心G点的能量依次为0,-0.168,-0.169 eV,计算(公式参见文献[15])得出自旋-轨道耦合分裂能Δso为-0.011 eV,晶体场分裂能Δcf为-0.158 eV,实验值[16]Δso约为0 eV,Δcf为-0.13 eV,两者接近.能带图显示Cu AlS2具有直接带隙结构,价带顶和导带底都位于布里渊区的G点处.计算的禁带宽度Eg为1.93 eV,尽管采用了广义梯度近似(GGA),但与实验值3.4 eV[7]和3.50 eV[4]相比偏小,这主要是由于GGA近似存在计算值偏低的普遍性问题.
从图3可看出,上价带主要由Cu 3 d,S 3p组成,还有少量的Cu 3 p,Al 3 p,Al 3s,下价带主要由S 3 p,Cu 3d,Al 3 s,Al 3p组成,还有少量的Cu 4s,Cu 3 p.对于-14.5 eV到-12.8 eV的价带,主要由S 3 s,Al 3p和Al 3 s组成,与其他2个价带之间的相互作用较弱.从态密度看CuAlS2的导带比较分散,主要由Al 3 p,Al 3s,Cu 3p,Cu 4s以及少量的S 3 p组成.价带顶和导带底分别取决于Cu 3d和Al 3 p,上价带主要由Cu 3 d组成.由于Cu原子之间d-d轨道相互作用,产生更高的价带态,表现出较强的非局域性,有利于空穴的迁移.从图2也可以看出价带顶附近能量起伏较大,空穴具有较小的有效质量.这也是Cu AlS2容易表现出p型导电以及容易实现p型掺杂的一个重要原因.
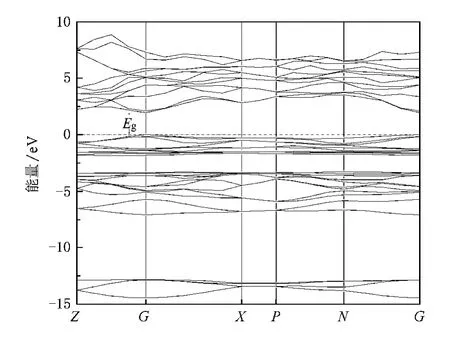
图2 CuAlS2的能带结构
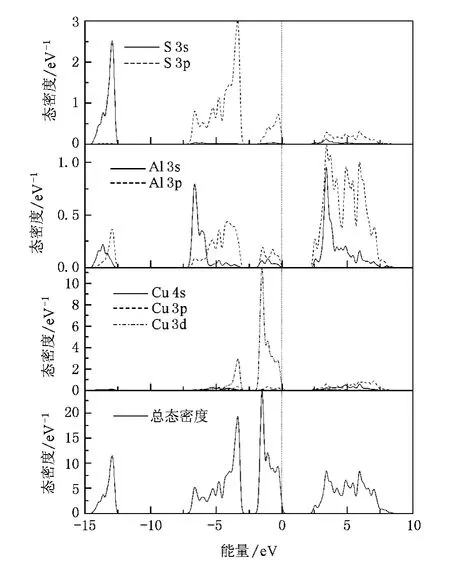
图3 CuAlS2的态密度分布
4.CuAlS2掺杂的计算
4.1.晶体结构
对非掺杂CuAlS2以及Mg和Zn掺杂CuAlS2模型进行了几何结构优化,优化后的晶胞参数见表1,其中实验值取自文献[9,10].从表1可以看出,Mg和Zn掺杂均使晶格常数增大,对于CuAlS2∶Mg,a增大0.25%,c增大0.35%;对于Cu AlS2∶Zn,a增大0.38%,c增大非常小.晶胞体积分别增大0.84%和0.77%.由于Mg和Zn的离子半径分别为0.072和0.074 nm,比Al离子半径0.053 nm大,且Mg的电负性较弱,在置换Al后与S形成了较强离子性的共价键,形成的Mg—S和Zn—S键的键长比Al—S的键长大,从而增大了晶格常数.Mg和Zn掺杂相比较,掺Mg后晶格常数c相对增大更多,晶胞体积增加程度更大.从能量上看,掺杂后的体系总能比未掺杂体系的总能低,这是由于Mg和Zn的化学势比Al的化学势更低,导致掺杂后体系总能降低.值得注意的是,Zn掺杂体系总能较Mg掺杂体系总能更低,说明Zn占据Al位置形成的ZnAl缺陷较Mg占据Al位置形成的MgAl缺陷更稳定.实验也证明了这一点,CuAlS2中Mg掺杂率在0—6%之间能保持黄铜结构,当Mg掺杂率为8%时,体系就会出现CuxS杂质相,表现出不稳定性,而Zn掺杂率在0—10%之间体系仍保持黄铜矿结构[9,10].

表1 CuAlS2,CuAlS2∶Mg,CuAlS2∶Zn晶胞参数和总能
4.2.电子结构
图4—8分别为Cu AlS2∶Mg和CuAlS2∶Zn掺杂情况的能带结构和分波态密度分布.由此可以看出,两种情况下的能带结构和态密度十分相似,电子态都发生了简并.
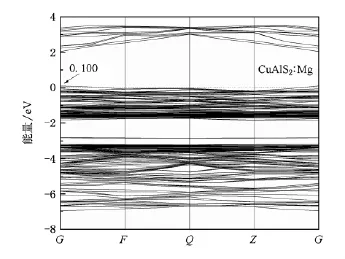
图4 CuAlS2∶Mg的能带结构
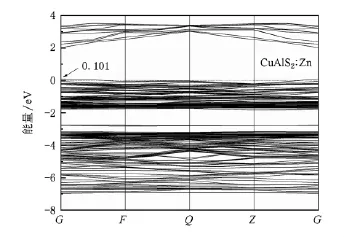
图5 CuAlS2∶Zn的能带结构
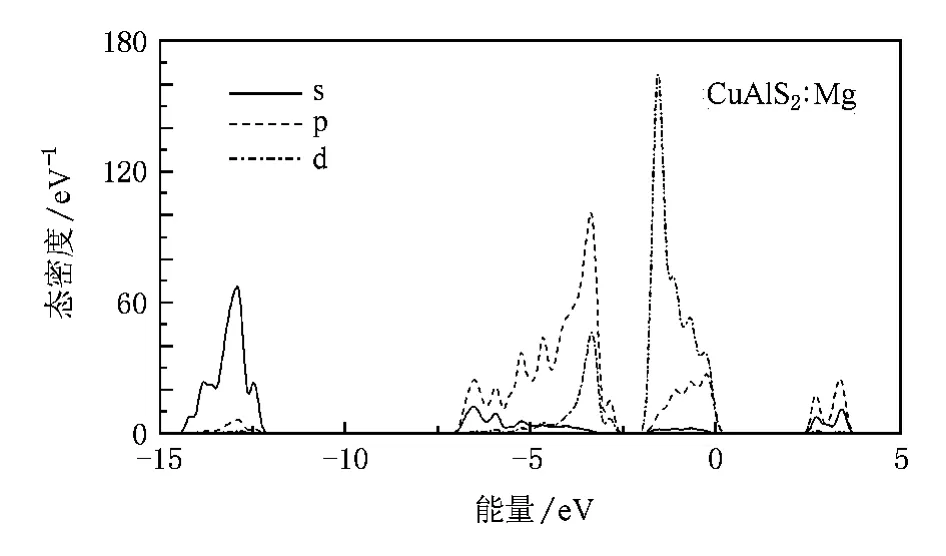
图6 CuAlS2∶Mg的分波态密度
掺杂Cu AlS2的能带结构显示费米能级进入价带顶,形成简并态,如图4和图5所示.掺杂使Cu AlS2价带出现多余的空穴载流子,在费米能级附近引入受主能级.从掺杂后的分波态密度(图6和图7)可以看出费米能级附近的态密度主要来源于d和p轨道,受主能级是杂质原子和Cu 3 d以及S 3 p作用的结果,而不是杂质原子直接形成的能级.经分析可知,杂质原子Mg和Zn均未在价带顶和导带低附近直接引入明显的电子态,这一点从Mg和Zn掺杂分态密度图上价带顶和导带底相似程度也可以看出.
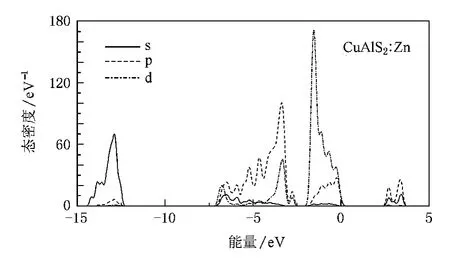
图7 CuAlS2∶Zn的分波态密度
从分波态密度图可以看出,Mg和Zn的掺杂都使导带的局域性更强,导带底稍稍向高能端移动,导带底分别为2.046和2.025 eV.这可能是由于Mg和Zn替位Al以后,Al原子减少,Al 3 p和Al 3s轨道之间以及与Cu 3p和Cu 4 s相互作用减弱,使得态密度分布变得更集聚.此外,掺杂后晶格常数变大,各轨道之间杂化减弱以及掺入二价元素后体系费米能级降低等,也可能是导致导带局域性增强,导带底抬高的原因.
Mg和Zn掺杂CuAlS2两个受主能级分别位于费米能级之上0.100和0.101 eV处,表明Mg占据Al位置形成的受主能级更容易电离产生空穴,Mg比Zn掺杂CuAlS2更容易实现p型电导,但两者相差并不是太大.这种结果与实验[9,10]符合得较好,Mg掺杂率在4%—6%之间,Zn掺杂率在2%—10%之间均能形成良好的p型电导,体材料电导率均大于5 S·cm-1,掺杂率同为6%时,体材料CuAlS2∶Mg的电导率为41.7 S·cm-1,CuAlS2∶Zn的电导率为18.8 S·cm-1.
4.3.差分电荷密度分布
图8为Cu AlS2及Mg和Zn掺杂CuAlS2晶面(110)的差分电荷密度分布图.由于掺杂原子外围原子的差分电荷密度变化不大,所以此处只局部显示掺杂原子及附近几个原子的差分电荷密度.由图8可看出,在未掺杂和掺杂情况下,原子间的成键性质差异较大,原子间的相互作用也不同,体系中的电荷发生了重新分配.从图8(a)可以看出,对于未掺杂的CuAlS2,Al和S,Cu和S之间形成了包含离子键成分的共价键,原子周围的差分电荷密度显现具有方向性的共价键特征.与Cu相比,Al的电负性更小,Al周围的电子更多地转移到S周围,Al—S键包含的离子键成分更多.从图8(b)可以看出,当Mg置换Al后,由于Mg的电负性小,Mg和S成键的离子性较强,对比图8(a)和(b)中S周围的差分电荷密度分布也可以看出,掺入Mg后S原子周围差分电荷密度分布更均匀,Mg—S键离子性更强.图8 (b)中Mg和S之间电荷密度增幅较图8(a)中Al和S之间的要弱,所以,Mg—S与Al—S相比成键强度相对较弱.从图8(c)可以看出,当Zn原子置换Al原子后,虽然Zn与Al的电负性相当,但Zn与S之间形成了较强离子键成分的共价键,成键强度比Mg—S键稍弱.从图8中S原子周围的差分电荷密度分布也可以看出,图8(c)中的分布最均匀,Al,Mg,Zn和S成键时,Zn—S键的离子性最强.
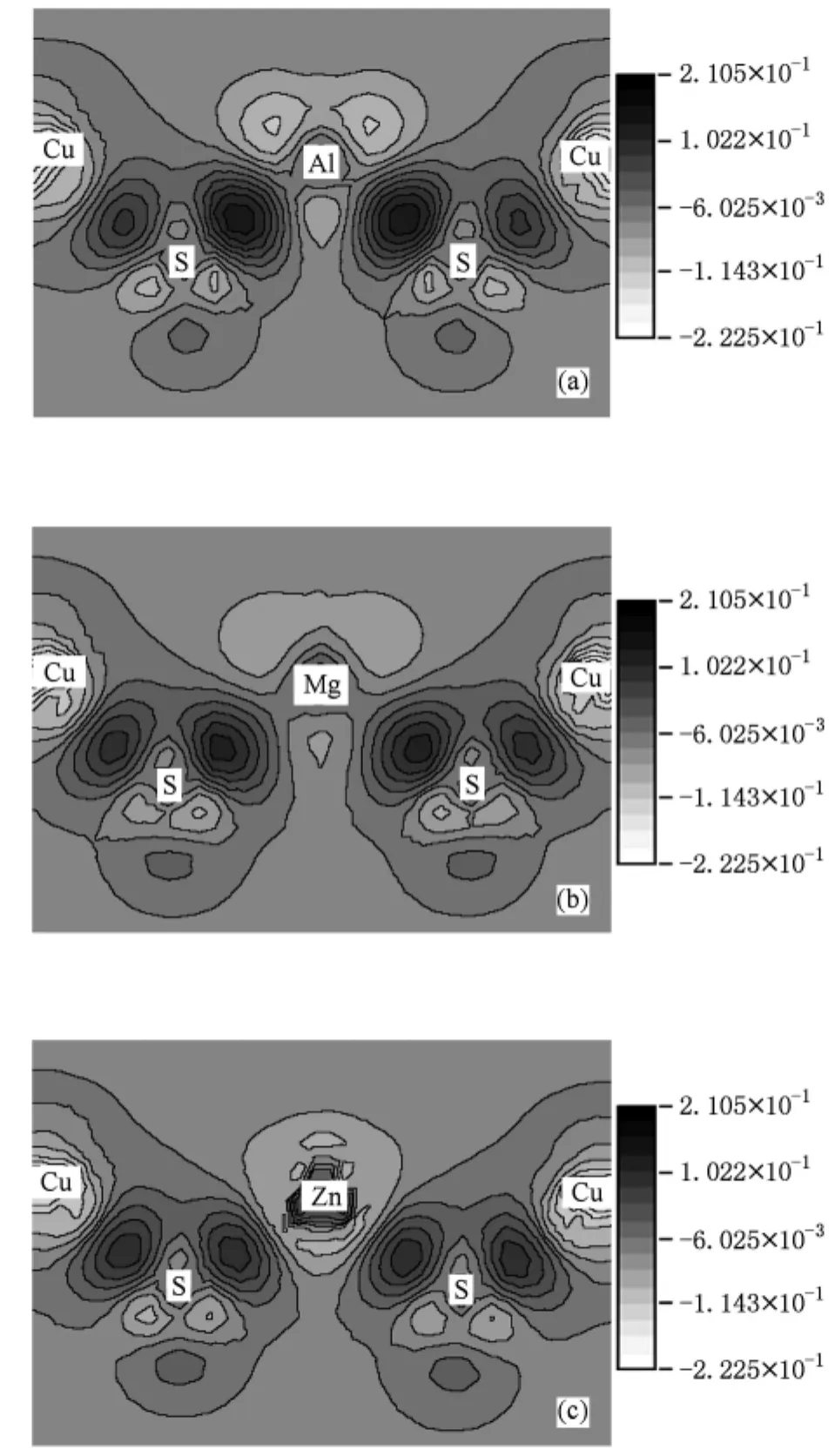
图8 (110)晶面的差分电荷密度分布(a)CuAlS2; (b)CuAlS2∶Mg;(c)CuAlS2∶Zn(单位:e/3)
5.结论
通过对黄铜矿结构CuAlS2及掺杂Cu AlS2∶Mg,Cu AlS2∶Zn电子结构的分析表明:Mg和Zn替位Al掺杂Cu AlS2晶格常数a和c均增大,晶胞体积分别增大0.84%和0.77%;与Al—S键相比,杂质原子与S原子成键的离子性增强,键长增大,成键强度相对减弱;Zn掺杂体系比Mg掺杂体系的总能更低,晶格结构较Mg掺杂体系更稳定;Mg和Zn掺杂都在Cu AlS2的价带顶引入受主态,Mg和Zn替位Al掺杂Cu AlS2均能形成良好的p型电导;Mg掺杂的受主能级具有更小的电离能.Zn与Mg掺杂Cu AlS2相比,两者均能引入受主态,受主能级电离能相差不大,而前者导致晶格畸变更小,掺杂体系更稳定.
[1]Brik M G 2009 J.Phys.:Condens.Matter 21 485502
[2]Li J,Zhu J 2007 Acta Phys.Sin.56 574(in Chinese)[李健、朱洁2007物理学报56 574]
[3]Feng J,Xiao B,Chen J C 2007 Acta Phys.Sin.56 5991(in Chinese)[冯晶、肖冰、陈敬超2007物理学报56 5991]
[4]Bhandari R K,Hashimoto Y,Ito K 2004 Jpn.J.Appl.Phys.Ⅰ43 6890
[5]Kuroki Y,Okamoto T,Takata M,Osada M 2006 Appl.Phys.Lett.89 221117
[6]Brini R,Schmerber G,Kanzari M,Werckmann J,Rezig B 2009 Thin Solid Films 517 2191
[7]Liu M L,Huang F Q,Chen L D 2008 Key Eng.Mater.368—372 666
[8]Liu M L,Wang Y M,Huang F Q,Chen L D,Wang W D 2007 Script.Mater.57 1133
[9]Liu M L,Huang F Q,Chen L D 2008 Script.Mater.58 1002
[10]Liu M L,Huang F Q,Chen L D,Wang Y M,Wang Y H,Li G F,Zhang Q 2007 Appl.Phys.Lett.90 072109
[11]Yang M,Wang Y H,Li G F,Shi Z,Zhang Q 2009 J.Vac.Sci.Technol.A 27 1316
[12]Milman V,Winkler B,White J A,Pickard C J,Payne M C,Akhmatskaya E V,Nobes R H 2000 Int.J.Quantum Chem.77 895
[13]Brandt G,Rauber A,Schneider J 1973 Solid State Commun.12 481
[14]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002 J.Phys.:Condens.Matter.14 2717
[15]Shirakata S,Chichibu S 1996 J.Appl.Phys.79 2043
[16]Shay J L,Tell B,Kasper H M,Schiavone H M 1972 Phys.Rev.B 5 5003
PACS:71.20.Nr,71.20.-b,61.72.U-,68.55.Ln
*Project supported by the Science and Technology Research Program of Guangdong Province,China(Grant No.A1100501).
Corresponding author.E-mail:phrhyao@scut.edu.cn
Electronic structure of CuAlS2doped with Mg and Zn*
Wan Wen-Jian Yao Ruo-HeGeng Kui-Wei
(School of Electronic and Information Engineering,South China University of Technology,Guangzhou 510640,China)
(Received 29 May 2010;revised manuscript received 8 October 2010)
Electronic structure of chalcopyrite-type Cu AlS2is analyzed from the band structure and the density of states.Compared with bulk CuAlS2,the effect of doping with Mg and Zn are investigated from the crystal structure,band structure,density of states and electron density difference.The results reveal that Mg and Zn substituting Al both increase the lattice constants and cell volume,yield acceptor states on the top of valence band,providing p-type conductance.CuAlS2∶Zn has a slightly larger ionization energy than Cu AlS2∶Mg,while the former has lower total energy and its crystal structure is more stable.
Cu AlS2,p-type doping,electronic structure,band structure
*广东省科技计划(批准号:A1100501)资助的课题.
.E-mail:phrhyao@scut.edu.cn
猜你喜欢
装备维修技术(2021年36期)2021-10-25
小天使·聪聪画刊(2021年2期)2021-09-10
弹箭与制导学报(2021年3期)2021-07-30
汽车零部件(2020年10期)2020-11-09
汉语世界(The World of Chinese)(2019年6期)2019-09-10
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
无机化学学报(2014年5期)2014-02-28
