
Ni4NdB电子结构和磁性能第一性原理研究*
2011-11-02 03:26:28易勇丁志杰李恺唐永建罗江山
物理学报 2011年9期
易勇丁志杰李恺 唐永建 罗江山
Ni4NdB电子结构和磁性能第一性原理研究*
易勇1)2)丁志杰1)2)李恺2)唐永建2)罗江山2)
1)(四川省非金属复合与功能材料重点实验室,省部共建国家重点实验室培育基地,绵阳621010)
2)(中国工程物理研究院,激光聚变研究中心,绵阳621900)
(2010年8月2日收到;2010年9月29日收到修改稿)
采用第一性原理,在局域自旋密度近似LSDA及LSDA+U近似,对Ni4NdB化合物进行结构优化,计算体系晶格常数,电子结构和磁性能.结果表明,Ni4NdB为带隙很小的金属导体,存在Nd-Ni铁磁耦合,体系总磁矩由Nd原子局域磁矩提供.体系原子成键较为复杂,Nd原子与近邻Ni原子成金属键,Nd原子与近邻B原子成较强离子键,Ni原子与近邻Ni原子间存在间接交换相互作用.在U作用下,体系磁矩与Nd原子磁矩变化一致,Ni原子磁矩在2.75 eV呈现磁有序-磁有序崩溃转变.
密度泛函理论,电子结构,磁性能,稀土过渡金属间化合物
PACS:75.50.Cc,71.20.Eh,71.15.Mb
1.引言
稀土碳硼、氮化合物[1—3]、稀土过渡族金属间化合物[4—7]等在实验研究和理论研究方面深受国内外研究学者关注.理论模拟研究发现稀土硼化物磁性能优异,主要源于高度局域的4 f电子,应用覆盖金属、半金属到半导体.由于稀土元素Re具有高度关联的4 f电子和5 d电子,过渡族元素T具有关联3 d局域电子,因此应用传统密度泛函理论很难处理这种强关联局域电子作用,从第一性原理的角度上,通常采用在位库仑排斥势修正(LSDA+U)、广义梯度近似(generalized gradient approximation,GGA)、自相互作用修正(self-interaction correction,SIC)、自能修正(SEC或GWA)等[8—10].磁性六方Ni4NdB是一种稀土过渡族金属间化合物,在磁性材料的实验研究中有重要意义,特别是非晶纳米晶软磁材料的研究.目前相关研究还鲜有报道,本文考虑自旋极化,采用LSDA及LSDA+U对六方Ni4NdB体系的电子结构和磁性进行了研究,以期对未来Ni4NdB的实验研究和应用进行探索和指导作用.
2.计算模型及方法

图1 六方Ni4NdB晶体结构
Ni4Nd B空间群为P6/mmm,为层状结构,如图1所示,其晶格参数为a=5.043,b=5.043,c= 6.941,α=90°,β=90°,γ=120°.本文首先采用LSDA近似对体系进行结构几何优化,将优化结构在LSDA和LSDA+U近似下对六方Ni4NdB体系电子结构和能态密度进行计算,在Hubbard模型一级近似下,考虑同一原子上自旋相反的局域电子在位(on-site)库伦排斥势U,以修正Ni原子3 d态巡游性强的强关联和分离费米面附近电子分布.Ni原子电子组态为[Ar]3d8,Nd原子电子组态为[Xe]4 f3,采用从头算全电子投影缀加平面波赝势法(projector augmented wave potentials,PAW)描述电子-离子相互作用,模拟采用5×5×4网格(k-points)对布里渊区进行积分,模拟平面波截断能Ecut=550.00 eV,smearing取0.2 eV,优化收敛精细度为2.0×10-6eV/atom.全部模拟计算在曙光集群上通过MedeA2.5实现.
3.结果与讨论
3.1.结构优化
首先采用LSDA近似优化体系结构,表1为晶格常数计算值与实验值的比较.对比可以发现理论模拟结果与实验值[11]一致性很好,晶格常数误差约为1.77%,说明该理论方法计算的结果具有较高的可靠性.

表1 Ni-Nd-B晶格常数优化计算值与实验值
3.2.LSDA近似下Ni4NdB态密度DOS及能带结构
图2为LSDA近似下对体系优化结构所得到的六方Ni4Nd B能带结构和对应体系(total density of states,TDOS),为了研究方便截取完整能带结构费米面附近的G-A矢量作为研究对象.图2(b)中费米面处多数自旋态与少数自旋态DOS分布均为非零,且对应图2(a)能带结构,费米能级与能带存在相交现象,说明六方Ni4NdB体系表现金属性.Alexandrov和Kaye在研究YBCO高温超导氧化物时引入电子比热计算关系[12]
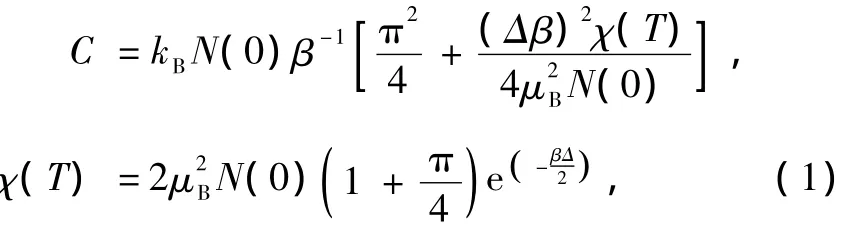
式中β=1/(kBT),在绝热近似下,由费米能级处体系基态单电子占据最高轨道和最低未占据轨道能隙Δ,及对应TDOS电子态密度数N,推算得出体系电子比热系数为3.20×10-6J/K2·mol,说明六方Ni4NdB为Kondo系统而非重费米子系统,从而为后续Ni基非晶软磁材料的低温电阻和体系磁性能的来源研究做铺垫.
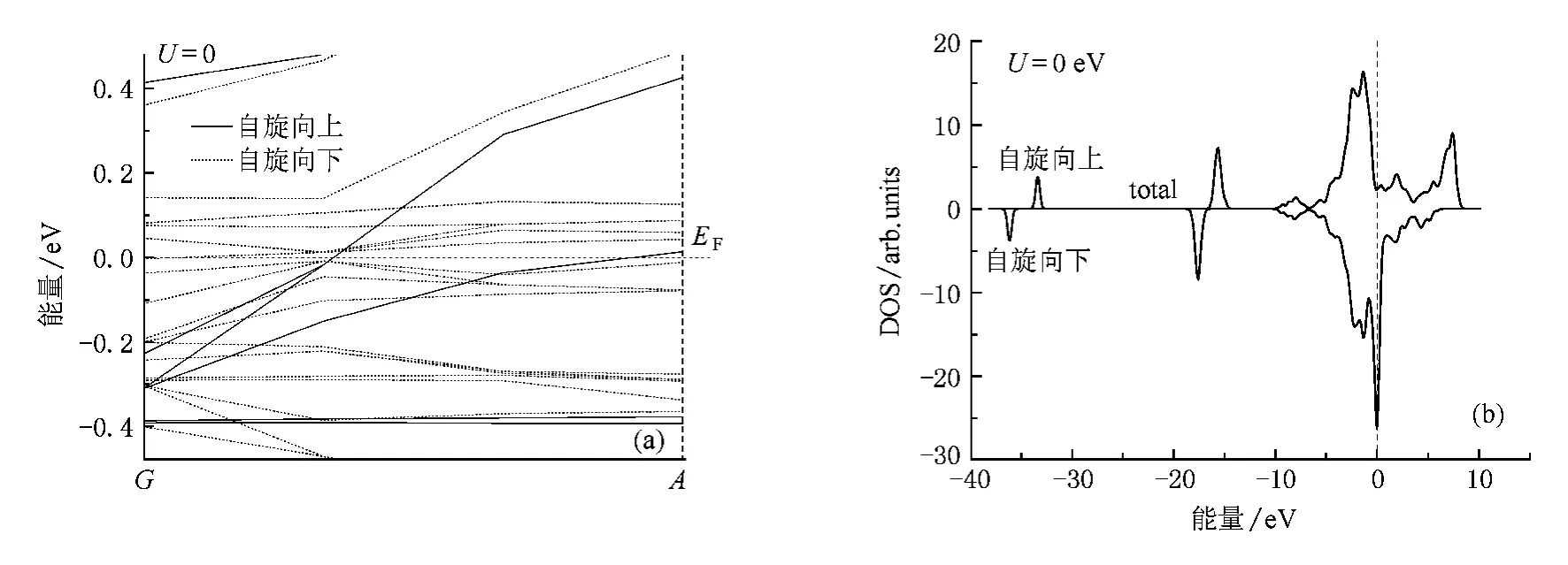
图2 (a)六方Ni4NdB费米能级附近能带结构;(b)TDOS
体系费米面附近带隙主要由自旋少数能带(minority band structure)提供.结合图3,Ni4NdB费米面附近价带主要由Nd-4 f构成,导带主要有Ni-3 d构成,带隙宽度为0.0493 eV,费米能级在带隙中间以下-0.0032 eV靠近价带处,体系显导体金属性,与TDOS图结果一致,由此可见Ni4NdB晶体是一种带隙很小的导体.
PDOS的计算对于研究体系电子局域性质及杂化特征等具有重要意义,图3为在计算TDOS的基础上对体系进行PDOS分析,根据Ni4Nd B导带DOS曲线,可以发现Ni-3 d为体系中主要传导电子,此外体系原子s,p电子也参与导电.结合图2(b)TDOS图,费米面附近DOS主要由Nd-4f态提供(约78.26%),结合图2(a),费米面处Nd-4 f态电子高度局域.能带主要分三段,其中在-37.12 eV到-32.45 eV区主要由s(Nd-6s态轨道电子)电子提供,Nd-6s态与Ni-3 p态轨道电子DOS曲线在少数自旋态部分存在重叠而多数自旋态部分不存在重叠现象,说明原子间存在相互作用但未成键,结合电荷密度(001)面分布(图4(a)),Ni与紧邻Nd间电子云少量交叠,存在s-p杂化.自旋极化引起Nd-6 s态发生自旋劈裂,交换劈裂能约0.87 eV,处于价带区,此外Nd-6s态电子在该区域具有一定局域性.-18.97 eV到-14.11 eV区域主要为p(Nd-5 p电子)电子贡献,B-2 s态和B-2 p态DOS曲线平缓,电子离域性很强,与Nd-5p态电子DOS相似且能带走向一致,结合图4(b),Nd与紧邻B原子明显存在电子云交叠而且重叠区域较广,故认为存在离子键作用,且成键很强.此外,Nd-5 p态与Ni-3 p态、Ni-4 s态存在较弱杂化,由于自旋极化的存在,引起p电子自旋分裂,Ni-3p,Ni-4 s电子因杂化有向少数自旋态迁移现象.-10.65 eV到9.06 eV区域附近f(Nd-4 f态电子)电子为主要贡献,Nd-4 f态电子几乎未参与杂化和电子波函数交叠,这主要是由于高能量Nd-4 f电子深埋在5s2p66 s2轨道电子内部,DOS分布在费米面两侧,峰形尖锐,能带带宽很窄,电子局域性很强.费米面低能部分电负性较大的B-2 s态与Ni-3 p态、Ni-4 s态PDOS存在包含关系,DOS间发生明显“共振”,形成分子轨道,其中B-2s态为成键分子轨道,并且贡献Ni-3 p态、Ni-4s态的反键分子轨道.B-2p态与Ni-3p态、Ni-4 s态在约-0.54 eV处DOS发生“共振”,亦形成分子轨道.费米面附近,Ni-3 d态与Nd-5d态DOS分布相似,存在较强3d-5 d杂化.Nd-4 f态在价带区自旋反向劈裂(少数自旋电子数多于多数自旋电子数),Ni-3p态、Ni-4 s态、Nd-5 d态、B-2 s态、B-2p态发生相对较弱的自旋正向劈裂,相比之下,反向劈裂程度比正向劈裂的和还要大得多,这种反向劈裂与正向劈裂引起Ni4NdB自旋能级劈裂,从而引起体系自旋对称性破缺,这是体系铁磁性的主要根源,计算得到其磁矩约0.170508μB.

图3 LSDA近似下六方Ni4NdB的PDOS

图4 LSDA近似下六方Ni4NdB100的(100)面(a)和(002)面(b)电荷密度图
图4 为Ni4Nd B在LSDA近似下的平均电荷密度分布图,由于体系结构的特殊性,切面选择(100)面分析Ni,Nd原子的电荷分布,(002)面分析Nd,B原子的电荷分布.结合电荷密度分布和DOS图相分析体系的成键和杂化情况:图4(a)中Nd原子失去电子与近邻两个Ni原子间成键明显,金属键为主; Nd原子与近邻Nd原子间存在杂化作用,该杂化类型通过间隙Ni原子形成间接交换相互作用,为4 f-5d-3 d-5 d-4f杂化作用形式;近邻Ni原子间电子波函数未发生重叠,无杂化和成键存在.图4(b)中Nd原子为明显失电子原子与两个近邻B受电子原子形成较强离子键,Nd原子轨道充当反键分子轨道,B原子轨道充当成键分子轨道,一个Nd原子与两个B原子形成一个分子轨道(与图3结果一致).
LSDA近似下得到Ni原子磁矩约为0.2μB,Nd原子磁矩为0.02μB,总磁矩约为0.170508μB,体系呈现铁磁耦合状态,这与实验中过渡金属元素子晶格-轻稀土元素子晶格形成铁磁耦合相符合[13],共有Nd-5 d电子的自旋传递交换作用使得Nd-4 f电子和Ni-3 d电子自旋动量方向相同,形成3 d-5 d杂化类型.
3.3.U值的影响
通过前面对优化体系在LSDA近似下的计算,发现体系中存在着较复杂的杂化关系、成键关系以及Nd-4 f的屏蔽效应.为了进一步研究Hubbard U对体系Ni-3d电子强关联作用的影响,固定交换积分JNi=1 eV[14],研究不同U值对体系DOS等的影响,并利用Mulliken布居分析得到电子轨道占据数,多数自旋态和少数自旋态电子占据数之差可以得到磁矩.

图5体系磁矩与Hubbard U的关系
图5 为不同U值下体系磁矩的变化,曲线总体上变化不大,可以从图上看到体系磁矩主要由Nd原子磁性提供,总磁矩变化趋势与Nd原子磁矩的变化趋势一致,所以在位库伦势U对Nd原子磁矩的影响比较大,Ni原子磁矩变化很小,表明Ni磁有序的程度比Nd磁有序要高得多,B原子磁矩总体为零,与非磁性原子的本质一致.总磁矩大多处于负值区主要是由于自旋引起能带反向劈裂引起的.当U为2 eV时,Ni磁矩达到最小值,约0.6μB,当U为2.75 eV时,体系磁矩为-3.64931μB,Nd局域磁矩为-3.76μB,Ni磁矩为0.16μB,几乎不对体系提供磁矩,Ni磁有序崩溃,说明U值对体系磁性的影响是不可忽略的.

图6 LSDA+U近似下Ni4NdB的TDOS和PDOS
图6 为在位库伦势为2.5 eV和8.0 eV时体系的TDOS和PDOS变化关系图,结合图3,Nd原子磁矩主要产生于被外层电子屏蔽的4f电子的高度局域性,4 f电子主要处在少数自旋占据态,由于在位库仑势和电子屏蔽的竞争作用,Nd-4f电子在U作用下呈现高度局域-局域减弱-高度局域的特性,磁矩变化较为明显(如图5).在费米面以下Nd-5 p和Nd-6s自旋态随U变化出现自旋劈裂-劈裂消失-自旋正向劈裂,且表现随着在位库仑势的增大分裂增大的趋势.具有巡游特性的Ni磁性产生于能带的劈裂,随着在位库伦势的变化,Ni能带先发生正向劈裂,之后劈裂减弱,最后又发生正向劈裂,自旋电子的运动可能是由于3 d-5 d杂化和在位库仑势的竞争作用所致,有利于体系获得较稳定的态结构,Ni磁矩对U值不是很敏感,自旋电子的运动很小.由于B无磁性,在位库仑势对其DOS电子分布影响不大.
4.结论
采用密度泛函理论(DFT)下的局域自旋密度近似LSDA及LSDA+U近似,考虑自旋极化下计算了新型稀土过渡族金属间化合物Ni4NdB的电子结构、能带结构及磁性特征等,通过分析得出以下结论:
1.在不考虑U作用下,体系磁矩主要由Ni原子提供.Ni4NdB体系显导体金属性,带隙很小约(0.0493 eV),费米面附近带隙主要由自旋少数能带提供,费米面附近靠近价带处DOS主要由Nd-4f态提供,靠近导带主要由Ni-3d态提供.体系比热系数为3.20×10-6J/K·mol.体系存在过渡金属元素子晶格-轻稀土元素子晶格形成的铁磁耦合.体系磁矩在考虑U作用下主要由Nd原子提供,Nd原子局域磁矩主要受到在位库仑势和3 d-5d杂化类型的影响,Ni原子局域磁矩在U为2.75 eV时发生磁有序崩溃,在位库仑势对Ni原子局域磁矩影响不是很明显.说明LSDA对于处理强关联Ni4Nd B体系的多体关联效应还有一定局限性,而采用LSDA+U修正则相对较为合理.
2.从电荷密度分布图中,证实Nd原子与近邻两个Ni原子以金属键作用为主.由于4 f-5 d-3d-5 d-4f杂化作用的存在,近邻Nd原子间呈现间接交换相互作用形式.Nd原子与两个近邻B原子以形成较强离子键的形式作用.
3.在研究强关联相互作用中,在在位库仑势的作用下,体系DOS变化明显.在位库伦势和电子屏蔽的竞争作用,发现Nd-4 f电子呈现高度局域—局域减弱—高度局域的磁特性,Ni能带劈裂变化出现正—负—正的特征.Nd磁矩变化形式对体系总磁矩的影响很明显.
[1]Larson P,Lambrecht W R L 2007 Phys.Rev.B 75 045114-1
[2]Anisimov V I,Zaanen J,Andersen O K 1991 Phys.Rev.B 44 943
[3]Leuenberger F,Parge A,Felsch W 2005 Phys.Rev.B 72 014427-1
[4]Severin L,Gasche T 1993 Phys.Rev.B 48 13547
[5]Oesterreicher H,Parker F T 1984 J.Appl.Phys.55 4334
[6]Zhang C W,Li H,Dong J M,Wang Y J,Pan F C,Guo Y Q,Li W 2005 Acta Phys.Sin.54 1814(in Chinese)[张昌文、李华、董建敏、王永娟、潘凤春、郭永权、李卫2005物理学报54 1814]
[7]Zhang J H,Liu S,Gu F,Yang L J,Liu M 2006 Acta Phys.Sin.55 2928(in Chinese)[张加宏、刘甦、顾芳、杨丽娟、刘楣2006物理学报55 2928]
[8]Gómez G,Cabeza G F,Belelli P G 2009 J.Magn.Magn.Mater.321 3478
[9]Maria Pugaczowa-Michalska 2008 J.Magn.Magn.Mater.320 2083
[10]P Jiji Thomas Joseph,Singh P P 2007 J.Magn.Magn.Mater.309 144
[11]Bilonizhko N S,Krik B I,Kuz'ma 1982 Akad.Nauk Ukr.RSR,Ser.B:Geol.,Khim.Biol.Nauki 21
[12]Alexandrov A S,Kaye G J 1999 J.Phys.:Condens.Matter 11 15
[13]He Zhiquan,He Wenwang 1990 J.Rare Eart.8 145(in Chinese)[黄智全、何文望1990中国稀土学报8 145]
[14]Cinquini F,Giordano L,Pacchioni G 2006 Phys.Rev.B 74 165403-1
PACS:75.50.Cc,71.20.Eh,71.15.Mb
*Project supported by the National Natural Science Foundation of China(Grant No.10476024).
E-mail:yiyong@swust.edu.cn
First-principles calculations of electronic structure and magnetism of Ni4NdB*
Yi Yong1)2)Ding Zhi-Jie1)2)Li Kai2)Tang Yong-Jian2)Luo Jiang-Shan2)
1)(State Key Laboratory Cultivation Base for Nonmetal Composite and Functional Materials,Mianyang 621010,China)
2)(Research Center of Laser Fusion of CAEP,Sichuan Mianyang 621900,China)
(Received 2 August 2010;revised manuscript received 29 September 2010)
The geometry optimization,the electronic and magnetic properties of the compound Ni4 NdB are studied by using firstprinciples within the local spin-density approximation(LSDA)and the LSDA+U approximation.The results indicate that the system is a metallic conductor with very small band gap,and that the total magnetic moment is provided by the local Nd magnetic moment.The system has very complex bonding,where Nd atoms and the neighboring Ni atoms form metal bonding,also Nd atoms and the neighboring B atoms form the strong ionic banding,besides Ni atoms and the neighboring Ni atoms forming an indirect exchange interaction.Under coulomb interaction,the system magnetic moment is consistent with that of the local Nd atom,and the collapse of magnetic ordering in 2.75 eV happens to the local Ni magnetic moment.
density functional theory,electronic structure,magnetic property,rare earth-transition metal compound
*国家自然科学基金(批准号:10476024)资助的课题.
E-mail:yiyong@swust.edu.cn
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
数学物理学报(2019年6期)2020-01-13 06:08:24
发明与创新·小学生(2019年12期)2019-12-05 06:02:28
发明与创新(2019年47期)2019-11-21 01:16:18
考试周刊(2018年39期)2018-04-19 10:39:44
深圳大学学报(理工版)(2015年6期)2015-11-26 12:33:48
深圳大学学报(理工版)(2015年5期)2015-02-28 16:21:26
河南科技(2014年23期)2014-02-27 14:18:52
