
RP-HPLC法测定替加环素的有关物质
2011-10-25 05:54吉同琴赵砚荣刘留成
药学研究 2011年12期
吉同琴,赵砚荣,魏 佳,刘留成
(江苏奥赛康药业股份有限公司,江苏南京211112)
替加环素(Tigecycline)为甘氨酰环素类广谱抗生素,其化学名为(4S,4aS,5aR,12aS)-9 -[2 -(叔 - 丁氨基)乙酰氨基]-4,7 - 双(二甲氨基) -1,4,4a,5,5a,6,11,12a- 八氢-3,10,12,12a-四羟基 -1,11- 二氧 -2 - 并四苯甲酰胺.替加环素的作用机制与四环素类抗生素相似,都是通过与细菌30s核糖体结合,阻止转移RNA的进入,使得氨基酸无法结合成肽链,最终起到阻断细菌蛋白质合成,限制细菌生长的作用.不过,替加环素与核糖体的结合能力要比四环素或米诺环素强5倍.2005年6月16日美国FDA批准注射用替加环素上市,商品名为Tygacil,规格为50 mg.批准的适应证为用于治疗患有复杂皮肤及软组织感染以及复杂的腹腔内感染的疾病[1].本文采用两种色谱条件测定其有关物质,方法1 参考《美国药典》30 版及《中国药典》2010 年版(二部)[2]盐酸米诺环素中有关物质检查项下的色谱条件建立,方法2参照文献[3]建立,通过对比研究,最后确定了采用方法1,其专属性强,能准确,快速检测替加环素的有关物质,能更好地控制替加环素的质量.
1 仪器与试药
LC-10AVP型高效液相色谱仪包括LC-10ATVP高压恒流泵和SPD-10AVP检测器(日本岛津公司);HW-2000色谱工作站(千谱软件);XS205型分析天平(Metter Toledo);PHS-3C型精密pH计(上海雷磁).
替加环素(批号:090801、090802、090803)、替加环素对照品(含量为99.6%,非水滴定法),均为本单位自制;四氢呋喃、N,N-二甲基甲酰胺(DMF)、乙腈、辛烷磺酸钠为色谱纯;水为超纯水;其余试剂均为分析纯.
2 溶液的配制
2.1 供试品溶液 取替加环素约25 mg,精密称定,置50 mL量瓶中,用流动相溶解并定容至刻度,摇匀,即得.
2.2 对照溶液 精密量取供试品溶液1 mL,置100 mL量瓶中,用流动相稀释至刻度,摇匀,即得.
3 色谱条件
3.1 方法1 色谱柱:以辛烷基硅烷键合硅胶为填充剂;流动相:0.2 mol·L-1醋酸铵溶液(含 0.01 mol·L-1乙二胺四乙酸二钠)-N,N-二甲基甲酰胺-四氢呋喃(67∶29∶4)(用三乙胺调 pH值至7.9);检测波长:254 nm;流速1 mL·min-1;进样体积:20 μL.
3.2 方法2 色谱柱:以十八烷基硅烷键合硅胶为填充剂;流动相:0.01 mol·L-1磷酸二氢钠缓冲液(含 0.02 mol·L-1辛烷磺酸钠及0.5%三乙胺,以磷酸调节pH值至3.0)-乙腈(75∶25);检测波长:246 nm;流速 1 mL·min-1;进样体积:20 μL.
4 方法与结果
4.1 系统适用性试验 取供试品溶液,按“3.1”项下色谱条件测定,典型色谱图见图1-1.供试品溶液色谱图中替加环素与相邻峰的分离度为3.4,替加环素的理论板数为3 446;取供试品溶液,按“3.2”项下色谱条件测定,典型色谱图见图1-2.供试品溶液色谱图中替加环素与相邻峰的分离度为3.3,替加环素的理论板数为2 785.
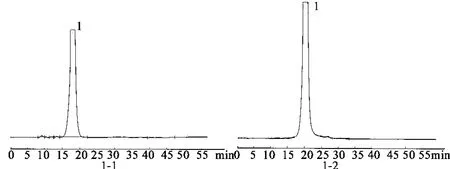
图1 替加环素样品色谱图 1.替加环素
4.2 破坏性试验
4.2.1 强酸破坏 取供试品溶液10 mL,加0.1 mol·L-1盐酸溶液1 mL,室温放置2 h,用1 mol·L-1氢氧化钠溶液调pH值至近中性,用0.45 μm 微孔滤膜滤过.按“3.1”项下色谱条件测定,色谱图见图2-A-1;按“3.2”项下色谱条件测定,色谱图见图2-A-2.
4.2.2 强碱破坏 取供试品溶液10 mL,加0.1 mol·L-1氢氧化钠溶液1 mL,室温放置2 h,用1 mol·L-1盐酸溶液调pH值至近中性,用0.45 μm 微孔滤膜滤过.按“3.1”项下色谱条件测定,色谱图见图2-B-1;按“3.2”项下色谱条件测定,色谱图见图2-B-2.
4.2.3 氧化破坏 取供试品溶液10 mL,加10%过氧化氢溶液1 mL,室温放置 10 min,用0.45 μm 微孔滤膜滤过.按“3.1”项下色谱条件测定,色谱图见图2 -C -1;按“3.2”项下色谱条件测定,色谱图见图2-C-2.
4.2.4 高温破坏 取供试品溶液10 mL,置100℃水浴中加热 1 h,放冷,用 0.45 μm 微孔滤膜滤过.按“3.1”项下色谱条件测定,色谱图见图2-D-1;按“3.2”项下色谱条件测定,色谱图见图2-D-2.
4.2.5 强光破坏 取供试品溶液10 mL,置7 500 lx光照强度下照射 2 h,用 0.45 μm 微孔滤膜滤过.按“3.1”项下色谱条件测定,色谱图见图2-E-1;按“3.2”项下色谱条件测定,色谱图见图2-E-2.

图2 破坏性试验样品色谱图
试验结果表明,本品在强酸、强碱、氧化、高温、光照等剧烈条件下均有不同程度的破坏,两个色谱条件均能检测在替加环素峰前后产生的数个降解产物峰,各降解产物峰与替加环素峰分离良好.
4.3 线性关系考察 取替加环素对照品48.56 mg,置50 mL量瓶中,用流动相溶解并定容至刻度,精密量取5mL,置50 mL量瓶中,用流动相稀释至刻度,再分别精密量取0.05、0.1、0.5、1.0、2.0 mL,置 5 个 10 mL 量瓶中,用流动相稀释至刻度,摇匀.取上述溶液,按“3.1”项下色谱条件测定.以替加环素溶液浓度(X)为横坐标,对应峰面积(Y)为纵坐标,进行线性回归,回归方程为:Y=879.46X+25.634,r=0.999 9结果表明,替加环素溶液浓度在0.49 ~19.42 μg·mL-1范围内与峰面积线性关系良好.
4.4 精密度试验 取4.856 μg·mL-1对照品溶液,按“3.1”项下色谱条件连续进样6次,替加环素峰面积的RSD为0.49%,结果表明,本法精密度良好.
4.5 稳定性试验 取供试品溶液两份,分别置于25℃和4℃环境中保存,按“3.1”项下色谱条件,分别于 0、1、2、3、4、5 h进样测定.结果表明,供试品溶液在室温条件下放置,杂质峰面积随着时间的增加而增加;在冷藏条件下3 h内有关物质无明显变化.说明该溶液不稳定,配制后应立即进样,必要时冷藏保存,不宜超过3 h.
4.6 检测限试验 取对照溶液,用流动相逐级稀释,以3倍信噪比计算检测限,按“3.1”项下色谱条件测定,替加环素的最低检测限为1.19 ng;按“3.2”项下色谱条件测定,替加环素的最低检测限为1.68 ng.
4.7 有关物质测定 精密量取供试品溶液、对照溶液各20 μL,分别注入液相色谱仪,记录色谱图至替加环素峰保留时间的2.5倍.供试品溶液色谱图中如有杂质峰,量取各杂质峰的峰面积和,并与对照溶液主成分的峰面积比较,用自身对照法计算有关物质的量,两种方法的测定结果见表1.

表1 替加环素有关物质的测定结果
5 讨论
5.1 检测波长的选择 称取替加环素适量,加流动相溶解并稀释制成15 μg·mL-1的溶液,经紫外分光光度计的扫描,替加环素于“3.1”项下的流动相中在254 nm和360 nm的波长处有最大吸收,结果见图3;通过HPLC双波长测定,两波长条件下,色谱峰个数相同,但在254 nm的波长下,替加环素以及有关物质的响应值较大,因此,选择254 nm作为检测波长.
5.2 起始原料及中间体与替加环素的分离度考察 称取9-氨基米诺环素盐酸盐(起始原料)、N-氯乙酰基-9-氨基米诺环素(中间体)适量,分别加流动相溶解并稀释成适宜浓度,按“3.1”项下色谱条件测定.再配制两者混合后加入替加环素的溶液,结果表明,起始原料、中间体均与替加环素峰分离度良好

图3 替加环素紫外吸收光谱图
5.3 流动相pH的选择 按“3.1”项下的流动相,调节pH值至7.8~8.0范围内,各杂质与替加环素峰均可以达到良好的分离效果.
5.4 小结 经过一系列方法学比较与验证,采用本文中“3.1”项下的色谱条件,各杂质峰与替加环素峰分离度良好,能准确地检测替加环素的有关物质,可用于替加环素的质量控制.
[1]Pankey GA.Tigecycline[J].J Antimicrob Chemother,2005,56(3):470 -480.
[2]国家药典委员会.中华人民共和国药典2010年版(二部)[S].北京:中国医药科技出版社,2010:697 -698.
[3]Li C,Sutherland CA,Nightingale CH,et al.Quantitation of tigecyline,a novel glycyclcycline[corrected],by liquid chromatography[J].J chromatogr B Analyt Techol Biomed Life Sci,2004,811(2):225 -229.
猜你喜欢
沈阳建筑大学学报(自然科学版)(2022年4期)2022-11-15
杭州电子科技大学学报(自然科学版)(2022年3期)2022-06-08
辽宁化工(2022年3期)2022-04-06
实用老年医学(2021年7期)2021-12-04
皮革制作与环保科技(2021年14期)2021-11-12
阅读(科学探秘)(2021年8期)2021-09-01
皮肤病与性病(2021年3期)2021-07-30
当代水产(2021年3期)2021-07-20
健康之家(2021年19期)2021-05-23
科学家(2021年24期)2021-04-25
