
纳米金的绿色合成与CTAB对金纳米聚集体的解聚集作用
2011-09-29 02:24:04戴兢陶杨锦明
无机化学学报 2011年2期
戴兢陶 朱 靖 杨锦明 沈 明*,1,
(1江苏省滩涂生物资源与环境保护重点建设实验室,盐城 224002)(2扬州大学化学化工学院,扬州 225002)(
3盐城师范学院化学化工学院,盐城 224002)
纳米金的绿色合成与CTAB对金纳米聚集体的解聚集作用
戴兢陶1,3朱 靖2杨锦明1,3沈 明*,1,2
(1江苏省滩涂生物资源与环境保护重点建设实验室,盐城 224002)(2扬州大学化学化工学院,扬州 225002)(
3盐城师范学院化学化工学院,盐城 224002)
室温下利用鞣酸(TA)既作还原剂又作保护剂,通过绿色还原方法制备了鞣酸包裹的金纳米粒子,并通过紫外可见光谱(UV-Vis)、透射电镜(TEM)、X射线衍射(XRD)、红外光谱(FTIR)等分别进行了表征和分析。结果表明,随着nTA/nHAuCl4比率的增加,紫外可见光谱显示鞣酸稳定的金溶胶最大吸收波长明显增大且吸收带变宽,而最大吸光率逐渐减小。TEM观察证实增加鞣酸用量会导致溶胶体系中花状金纳米聚集体的形成。实验发现,阳离子型表面活性剂十六烷基三甲基溴化铵(CTAB)对鞣酸保护的金纳米聚集体具有强烈的解聚集作用,在剧烈搅拌的条件下CTAB能使金纳米聚集体成功解聚为单分散状态的金纳米粒子,提高体系温度可明显促进CTAB对金纳米聚集体的解聚集作用。
鞣酸;金纳米粒子;绿色合成;十六烷基三甲基溴化铵;解聚集作用
0 引 言
金属纳米粒子由于受到表面效应[1]和宏观量子隧道效应[2]等因素的影响,在电子学[3]、磁学[4]、光学[5]、热学[6]和力学[7]等性质上明显不同于宏观金属块体。对于贵金属而言,其独特的物理、化学性质与纳米结构的特殊性能的有机结合,使其具有了异乎寻常的表面化学性质,从而在催化、能源、微电子和生物等领域有着广阔的应用前景,受到人们越来越广泛的重视[8-9]。
近年来,绿色合成贵金属纳米材料已成为纳米科技领域关注的热点。Sastry研究小组[10]用柠檬草提取液与三价金离子反应制备了三角状金纳米粒子,通过简单地改变反应介质中柠檬草提取液的浓度调控三角状金纳米粒子的尺寸,后续又研究了反应温度、卤素离子的加入等对绿色制备金纳米粒子形貌与尺寸的影响[11]。Kasthuria等[12]利用芹菜苷既作还原剂又作保护剂,采用绿色合成的方法制备出了各向异性的金纳米结构和准球形的银纳米粒子。尹晋津等[13]通过比较柠檬酸钠、柠檬酸钠-鞣酸、抗坏血酸和硼氢化钠等还原方法制备金溶胶,发现柠檬酸钠-鞣酸还原法制备金纳米粒子的过程较为简单,制得的金粒子可应用于检测生物物质的信号标记。Tian等[14]在水体系中采用种子生长法实现了鞣酸对硝酸银的还原,制备出尺寸较大的银纳米盘。郭慧尔等[15]用氨水调节溶液的pH值得到了鞣酸还原并保护的银纳米结构,若预先加入水溶性高聚物聚乙烯吡咯烷酮(PVP),产物中则会有银纳米线产生。本工作用鞣酸既作还原剂又作保护剂,通过绿色合成的方法制备了鞣酸稳定的金溶胶,并考察了鞣酸用量、反应温度和添加剂等对金纳米粒子尺寸及形貌的影响。实验发现,随着还原剂鞣酸用量的增加,所得金溶胶的紫外可见吸收出现明显红移,透射电镜观察证实此现象为金纳米粒子间产生聚集所致。通过添加适量阳离子型表面活性剂十六烷基三甲基溴化铵(CTAB),可成功实现对鞣酸保护的金纳米聚集体的解聚作用,并试图对解聚集机理进行合理的推测。
1 实验部分
1.1 试剂与仪器
鞣酸(C76H52O46,TA),HAuCl4·4H2O,十六烷基三甲基溴化铵(CTAB),聚乙烯吡咯烷酮K-30(PVP),十二烷基硫酸钠(SDS)等均为分析纯试剂,购自国药集团化学试剂有限公司。实验用水为二次蒸馏水。
UV-2501 PC紫外可见光谱仪(日本,岛津公司);TECNAI-12透射电镜(TEM,荷兰,菲利浦公司);740型红外光谱仪(美国,尼高力公司);电泳实验采用U型电泳管和LW30J3型直流稳压稳流电源(上海力友电气有限公司);用D8 Advance Superspeed X射线衍射仪(德国,布鲁克公司)进行材料结构的表征,测试中采用了 Cu 阳极靶(λ=0.15406 nm)、石墨单色器,工作电压为40 kV,工作电流为200 mA,扫描范围(2θ)为 20°~90°,扫描间隔为 2θ=0.04°。
1.2 鞣酸稳定的金溶胶制备
金纳米颗粒在常温下通过鞣酸还原氯金酸制得。 典型的实验如下,取 3.0 mL 1.62 mmol·L-1的鞣酸水溶液[nTA/nHAuCl4=1∶1(R)]置于 50 mL 烧杯中,再加入16.5 mL蒸馏水,在剧烈的磁力搅拌条件下加入 0.50 mL 9.71 mmol·L-1的氯金酸水溶液, 约经数秒钟溶液出现淡红色,随着反应进行体系颜色加深,数分钟后溶胶颜色基本稳定,继续搅拌约 0.5 h,由鞣酸还原并稳定的酒红色金溶胶 (标为溶胶A)即被制得并留待进一步处理和表征。固定HAuCl4含量和体系总体积,增加鞣酸用量分别至R为7∶3、10∶3和5∶1),重复上述实验,可分别制得紫红色(标为溶胶B)、蓝紫色(标为溶胶C)和蓝色金溶胶(标为溶胶D)。
1.3 CTAB对鞣酸保护的金纳米聚集体的解聚集作用
向上述制备的鞣酸还原并稳定的新鲜蓝色金溶胶D中添加阳离子型表面活性剂CTAB[0.177 0 g,nTA/nHAuCl4=200∶1(R1)],并在25℃条件下持续搅拌12 h,溶胶D的颜色逐渐由蓝色变为红色,提高体系温度则能明显加快金溶胶的变色过程。实验表明,CTAB对鞣酸保护的金纳米聚集体具有强烈的解聚集作用。若以阴离子型表面活性剂SDS(0.2801 g)代替CTAB进行实验,则会加剧粒子的进一步聚集,溶胶稳定性显著下降,而加入PVP(0.524 0 g)到溶胶D,体系仍保持蓝色,但稳定性大为提高。
2 结果与讨论
2.1 不同鞣酸用量对金溶胶紫外可见光谱及粒子形貌的影响
图1给出了不同R条件下经常温搅拌制备的4种金溶胶的紫外可见光谱和数码照片。由图1a可见,鞣酸保护的金纳米粒子展示了典型金溶胶所特有的光学特征,即最大吸收波长(λmax)在500~550 nm可见光区产生表面等离子共振吸收[16-17],如溶胶A的λmax≈533 nm[图1a中的曲线A]。粒子的尺寸效应通常可从金属溶胶的吸收光谱反映出来,即随着粒径的变小,粒子的表面等离子共振吸收将向高能量方向移动,而纳米粒子的尺寸增大、“絮凝作用”(聚集或凝聚)的产生或高轴径比(非球状)粒子的生成均会导致溶胶吸收光谱的红移[18-20]。增大鞣酸用量所得金溶胶 B、C和 D的 λmax分别为 550、561和584 nm(对应于图1a中的曲线B、C和D),显示金溶胶的最大吸收波长随鞣酸用量增加而明显增大且吸收带变宽,其最大吸光率则随鞣酸用量的增加而逐渐减小。图1b所示溶胶的数码照片也清楚地显示,随着鞣酸用量的增加,所制备的4种金溶胶的颜色从酒红色、紫红色到蓝紫色再到蓝色,这与溶胶的紫外可见光谱相一致。该现象与用微波辐射辅助的乙二醇还原法制备金纳米粒子的情形相象[21],即呈现出貌似“反常”的颜色递变现象,因为在一般情况下,体系中还原剂用量增加,反应速率会加快,导致金粒子的成核速率加快,理应得到更小尺寸的金纳米颗粒。实验过程也的确显示鞣酸用量的增加会加快反应的速率,反映在金溶胶体系的变色时间逐渐缩短。上述结果暗示,新鲜制备的4种金溶胶中,溶胶A的粒子尺寸较小,且单分散性较好,但溶胶B、C和D中的金纳米粒子尺寸可能较大或由于产生金粒子间的聚集现象而生成了较大尺寸的纳米聚集体。这一推论得到了TEM观察的进一步证实。
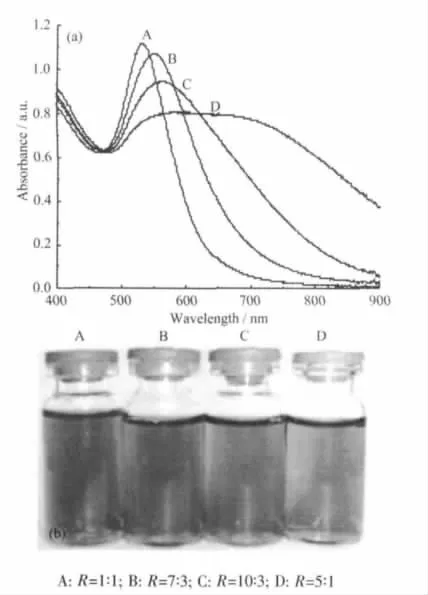
图1 不同nTA/nHAuCl4(R)时金胶体的紫外可见吸收光谱(a)和数码照片(b)Fig.1 UV-Vis absorption spectra(a)and digital photos(b)of gold colloids at different nTA/nHAuCl4ratios
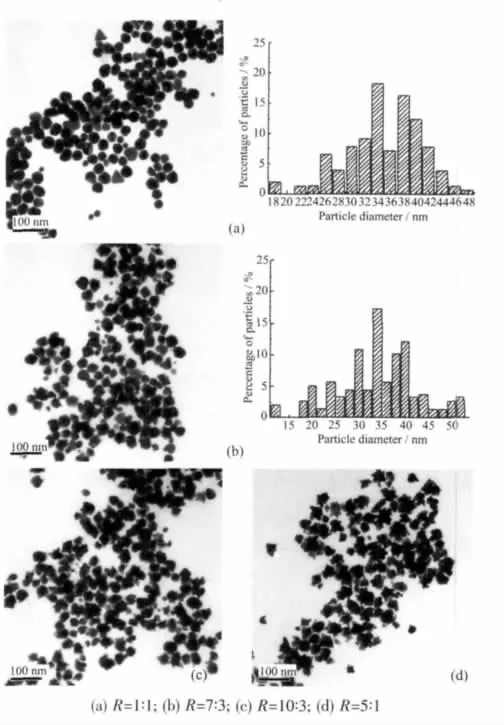
图2 不同nTA/nHAuCl4(R)时金纳米粒子的TEM照片和粒径分布直方图Fig.2 TEM micrographs and histograms of gold nanoparticles at different nTA/nHAuCl4(R)ratios
不同R条件下所得金纳米结构的TEM照片和粒径分布直方图如图2所示。当R=1∶1时,所得金纳米粒子大多为球形(约占94.2%),亦有极少量三角形貌的粒子存在,粒径分布直方图显示粒子的平均粒径和标准偏差为(34.6±9.1)nm(见图 2a),说明在此条件下所获得的金纳米粒子具有较好的单分散性;当R=7:3时,所制备的金纳米粒子呈不规则的似球形,因少数粒子的边缘聚集了小尺寸金粒子,导致粒子整体形貌不规则,其平均粒径和标准偏差为35.1±9.9 nm;当 R=10∶3 时,部分金粒子边缘因存在较多的小粒子而呈现似花状,这可能是金粒子间发生聚集所致(见图 2c),其平均粒径为 36.4 nm;当 R=5∶1时,鞣酸保护的金粒子总体呈现出不规则的花状形貌,其平均粒径为42 nm。结合溶胶D的紫外可见光谱(图1a中的曲线D)判断照片上的颗粒可能是金纳米粒子的聚集体。说明随着鞣酸用量增加,金纳米粒子总体尺寸并未显著增加,只是金粒子间发生了明显聚集。鞣酸是一种树枝状的大分子化合物,碳链较长,吸附于金粒子表面的鞣酸分子在一定程度上提高了金溶胶的稳定性,但其亲水支链上所带的羟基和羧基亦可能在金粒子碰撞过程中通过分子间的氢键或范德华力作用,拉近纳米金颗粒之间的距离,产生晶核碰撞融合,形成较大尺寸的颗粒来降低表面能或颗粒间彼此吸引形成局部团聚,造成颗粒的均匀性和分散性变差。当体系中鞣酸浓度增加时,这种通过鞣酸分子的“搭桥作用”所形成的金粒子聚集现象将更为显著。
2.2 反应温度对金溶胶紫外可见光谱及粒子形貌的影响
反应体系中温度变化往往对粒子的生成速率和形貌具有较大影响[22]。我们尝试了固定nTA/nHAuCl4(R=5∶1)和反应时间(30 min),在不同温度条件下(25、50、75和100℃)进行反应,所得金溶胶的紫外可见光谱见图3。由图可见,当反应温度为25℃时,其紫外可见光谱呈一平台状的宽带吸收,λmax为584 nm;当温度升高至50℃时,紫外可见光谱的λmax蓝移至571 nm,吸收谱带变窄,最大吸光率增加;随后反应温度分别升高为75和100℃,对应金溶胶的紫外可见光谱的λmax进一步蓝移至551和543 nm,同时对应吸收曲线的最大吸光率随温度升高而增加,吸收谱带逐渐窄化。这一结果暗示,在25~100℃范围内,随反应温度的提高,所得金纳米粒子尺寸有可能减小或粒子的聚集状态发生了改变。
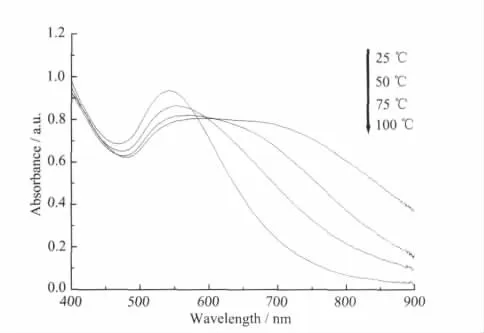
图3 不同温度下鞣酸稳定的金溶胶的紫外可见光谱Fig.3 UV-Vis absorption spectra of tannic acid-stabilized gold colloids at different temperatures
图4给出了不同温度时所得鞣酸包裹的金纳米结构的TEM照片。图4a显示出25℃时反应得到的金纳米聚集体呈现出不规则的花状形貌,其平均粒径为42 nm。从TEM照片上可清楚地看到较小尺寸的金纳米粒子附着在花状粒子的边缘,结合紫外可见光谱(图4a)判断照片上的花状粒子是由较小尺寸的金纳米粒子聚集而成;当温度升高到50℃时(图4b),似球形金纳米粒子和花状粒子共存,其平均粒径为27 nm;当温度继续升高到75℃时(图4c),似球形金纳米粒子数目增多,而似花状形貌的粒子数明显减少,粒子的平均粒径为21.1 nm;在沸腾状态下由鞣酸绿色合成的金纳米粒子则几乎全为似球形(图4d),粒子的平均粒径为 18.3 nm,且处于良好的单分散状态。从上述实验结果可以得出如下结论:一方面,温度升高,分子的热运动加剧,反应物分子的碰撞加剧,导致粒子的成核速率加快,生成的金纳米粒子尺寸减小;另一方面,温度升高,鞣酸分子对金纳米粒子的保护作用增强,使得金粒子间聚集程度明显降低,从而在一定程度上提高了金纳米粒子的单分散性。

图4 不同温度下鞣酸保护的金纳米粒子的TEM照片Fig.4 TEM micrographs of tannic acid-protected gold nanoparticless at different temperatures
2.3 外加保护剂对绿色合成金纳米粒子形貌及稳定性的影响
图5给出了25℃时向溶胶D中分别添加适量CTAB、SDS 和 PVP[nCTABorSDSorPVP/nHAuCl4=200∶1(R1)],剧烈搅拌12 h后所得到的3种溶胶的紫外可见光谱。在25℃条件下,向溶胶D中加入CTAB之后,溶胶会从蓝色逐渐变为红色,新溶胶的紫外可见光谱(图5a)显示其λmax为538 nm。CTAB加入到溶胶D中所导致的变色效应表明,原蓝色溶胶D中的金纳米聚集体已被CTAB成功解聚,而原溶胶D所呈现的蓝色应是溶液中金纳米粒子形成花状聚集结构的反映。TEM观察结果进一步证实了这一推论。
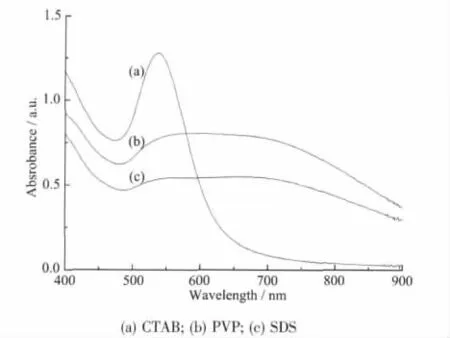
图5 溶胶D中外加保护剂后所得金溶胶的紫外可见光谱Fig.5 UV-Vis absorption spectra of gold colloids after adding some protecting reagents into colloid D
图6a的TEM照片中的金粒子呈现单个分散的似球状,其平均粒径和标准偏差为(30.4±6.4)nm,而较大尺寸的花状聚集体已不复存在,说明阳离子型表面活性剂CTAB的加入,对鞣酸包裹的花状金纳米聚集结构具有强烈的解聚集作用。实验还显示,倘若在加入CTAB后,剧烈搅拌的同时辅以常规加热或微波加热,均可大大缩短溶胶D的变色时间,亦即通过搅拌和加热均可促进CTAB对鞣酸保护的金纳米聚集体的解聚集作用。而加入水溶性高分子保护剂PVP后对溶胶D的紫外可见光谱的影响较小[见图5(b)],结合图2a中的曲线D可见,两条吸收曲线形状非常相似,在大于500 nm范围均出现一宽的吸收带,说明PVP不像CTAB分子那样对金纳米聚集体有解聚作用,但能显著提高溶胶D的稳定性,新溶胶经数周放置后不会发生任何沉淀。相反,当加入等物质的量SDS后则会使溶胶D的稳定性明显下降,其相应溶胶的紫外可见光谱见图5c,对应吸光率也有所降低,说明溶胶D中的粒子聚集程度进一步加剧,若将SDS处理过的新溶胶静置,则会很快失去稳定性,两天后金粒子就会全部沉淀下来。图6(b)、(c)分别显示了PVP和SDS加入溶胶D,经剧烈搅拌12 h后所得金纳米粒子的TEM照片,图中的金纳米结构与图2(d)所示的结果相似,即仍呈现聚集状态,说明花状粒子的形貌与未加PVP、SDS时相似,平均粒径分别为43[图6(b)]和64 nm[图6(c)],加入SDS后所致的粒子尺寸显著增加表明金粒子间的聚集程度加大。
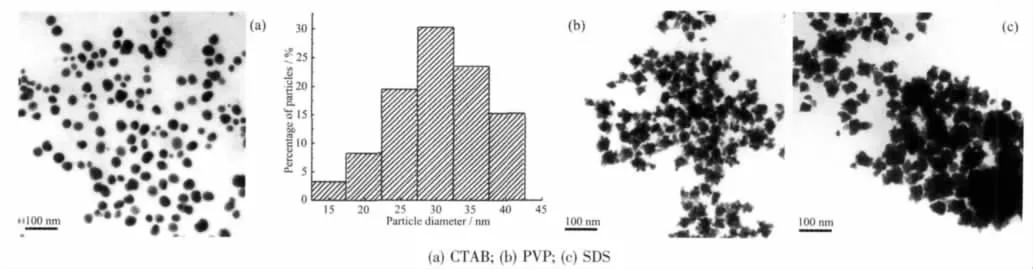
图6 25℃下溶胶D中外加保护剂所得金纳米粒子的TEM照片和粒径分布直方图Fig.6 TEM micrographs and histogram of gold nanoparticles after adding some protecting reagents into colloid D at 25℃
如前所述,随着nTA/nHAuCl4增加,所得金溶胶的紫外可见吸收呈现明显的红移现象,TEM观察显示,这一现象的发生是由于鞣酸用量增加所引起的,其鞣酸分子的“搭桥作用”导致了金纳米粒子聚集体的形成。当向产生明显聚集的蓝色金溶胶(如溶胶D)中添加阳离子型表面活性剂CTAB,经12 h搅拌后即能成功将蓝色溶胶转变成酒红色溶胶,进一步的实验表明,这一变色过程是由CTAB对金纳米粒子聚集体的成功解聚实现的。分析上述实验结果,我们拟用如图7的示意图表示CTAB对鞣酸保护的金纳米聚集体的解聚集作用,并试图做出如下解释:首先,根据鞣酸稳定的金溶胶的电泳实验发现金纳米粒子及其聚集体在电场作用下朝正极移动,表明鞣酸保护的金纳米粒子带负电,而在水溶液中CTAB可形成带正电的胶束,这样正、负粒子间的静电吸引作用有利于CTAB分子插入到金纳米聚集体的粒子之间;其次,CTAB分子与鞣酸分子在金纳米粒子表面会产生竞争性吸附作用,由于鞣酸分子的酚羟基对金纳米粒子的物理吸附作用较弱,因而带正电的表面活性剂CTAB分子在竞争吸附中占据优势;最后,溶液中足量的CTAB分子存在,可于金粒子表面形成亲水基朝向水介质而憎水链朝向憎水层的双层保护结构,从而使解聚后的金纳米粒子能稳定存在于水介质中。结合前面的实验结果,当加入阴离子型表面活性剂SDS时,则可能由于带负电的胶束对带同电性的金纳米聚集体的静电排斥作用,使得聚集作用加强,导致金溶胶更快失去稳定性;而亲水性高分子保护剂PVP的加入,不能对金纳米聚集体起到解聚集作用,但吸附于金纳米聚集体表面的PVP分子却显著提高了金溶胶的稳定性。
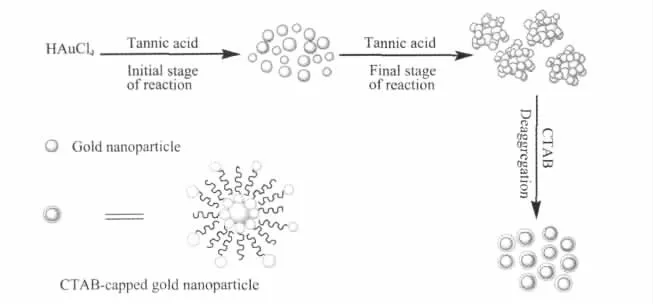
图7 鞣酸保护的金纳米聚集体形成和CTAB诱导的解聚集作用示意图Fig.7 Schematic diagram for the formation and deaggregation of tannic acid-protected gold nanoaggregates induced by CTAB
2.4 鞣酸保护的金纳米粒子的结构表征
图8是鞣酸(TA)和鞣酸稳定的金纳米粒子在500~4000 cm-1范围内的FTIR图谱。与鞣酸相比,-OH的伸缩振动在3379 cm-1附近显示出宽而强的吸收带,而鞣酸还原的金纳米粒子其对应-OH的伸缩振动吸收带移至3 402 cm-1附近,且吸收带变窄强度减弱,这说明金纳米粒子是通过酚羟基与Au3+反应而导致酚羟基数目减少。另外,1 614、1 535和1 449 cm-1为苯环的伸缩振动[见图8(a)],而图8(b)所示金纳米粒子中仅出现位于1 645和1 452 cm-1两个吸收峰,这是由于发生氧化还原反应后酚羟基氧对苯环供电子能力增加,使苯环碳架的吸收峰位置和个数发生了变化所致。
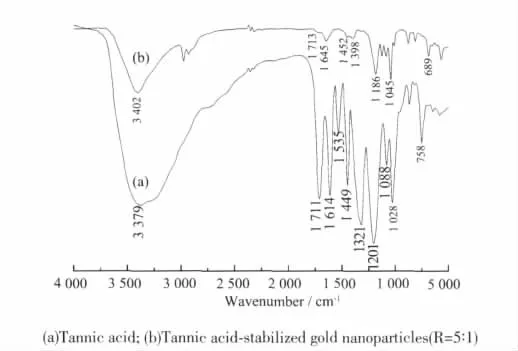
图8 鞣酸粉末与鞣酸保护的金纳米粒子的红外谱图Fig.8 FTIR spectra of pure tannic acid powder(a)and tannic acid-stabilized gold nanoparticles(b)
前述溶胶A和溶胶D两体系经处理后所对应样品的XRD图如图9所示。由图可见,由鞣酸包裹的两个粉末样品(样品A和D)均呈现出对应于大块面心立方(fcc)金晶体晶格参数的衍射峰(分别对应于图9a和b),只是由于粒子的尺寸效应使衍射峰明显变宽。
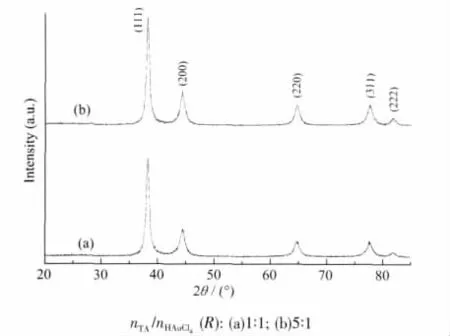
图9 鞣酸包裹的金纳米粒子的XRD图Fig.9 XRD patterns of tannic acid-capped gold nanoparticles
3 结 论
利用鞣酸作为还原剂通过绿色合成法制备了鞣酸保护的金纳米粒子。结果表明,在无其他保护剂的前提下,单独增加鞣酸用量,会导致金纳米粒子聚集形成花状纳米聚集体。电泳实验表明鞣酸保护的金纳米聚集体带负电,当向溶胶体系中加入阳离子型表面活性剂CTAB时,发现其对金纳米聚集体具有强烈的解聚集作用,而加入阴离子型表面活性剂SDS和水溶性高分子PVP均不具有解聚集作用,但PVP的加入可明显提高金溶胶的稳定性,相反SDS的加入则使金溶胶的稳定性显著下降。
[1]WANG Yong-Kang(王永康),WANG Li(王立).Science and Technology of Nanomaterials(纳米材料科学与技术),Hangzhou:Zhejiang University Press,2002.
[2]GU Ning(顾 宁),FU De-Gang(付 德 刚 ),ZHANG Hai-Qian(张海黔).Nanotechnology and Application(纳米技术与应用),Beijing:Posts and Telecom Press,2002.
[3]Simon U,Schon G,Schmid G.Angew.Chem.,Int.Ed.Engl.,1993,32(2):250-254
[4]Sun S H,Anders S,Hamann H F,et al.J.Am.Chem.Soc.,2002,124(12):2884-2885
[5]HUANG Yu-Ping(黄玉萍),XU Shu-Kun(徐淑坤),WANG Wen-Xing(王文星),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,123(10):1683-1688
[6]Roucoux A,Schulz J,Patin H.Chem.Rev.,2002,102(10):3757-3778
[7]ZHANG Zhi-Kun(张志焜),CUI Zuo-Lin(崔作林).Nanotechnology and Nanomaterials(纳米技术与纳米材料).Beijing:National Defence Industry Press,2000.
[8]JIANG Long(江龙).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2000,16(2):185-194
[9]Burda C,Chen X B,Narayanan R,et al.Chem.Rev.,2005,105(4):1025-1102
[10]Shankar S S,Rai A,Ahmad A,et al.Chem.Mater.,2005,17(3):566-572
[11]Rai A,Singh A,Ahmad A,et al.Langmuir,2006,22(2):736-741
[12]Kasthuria J,Veerapandianb S,Rajendiran N.Colloids and Surfaces B:Biointerfaces2009,68(1):5560
[13]YIN Jin-Jin(尹晋津),XU Li-Jian(许利剑),ZENG Xiao-Xi(曾晓希).J.Hunan University of Technology(Hunan Gongye Daxue Xuebao),2008,22(1):104-108
[14]Tian X L,Wang W H,Cao G Y.Mater.Lett.,2007,61(1):130-133
[15]GUO Hui-Er(郭慧尔),YAN Peng-Xun(闫鹏勋),LI Ming(黎明).Rare Metals Letters(Xiyou Jinshu Kuaibao),2007,26(11):31-34
[16]Aslan K,Perez-luna V H.Langmuir,2002,18(16):6059-6065
[17]Jana N R,Gearheart L,Murphy C.J.Langmuir,2001,17(22):6782-6786
[18]Weisbecker C S,Merritt M V,Whitesides G M.Langmuir,1996,12(16):3763-3772
[19]Mayya K S,Patil V,Sastry M.Langmuir,1997,13(15):3944-3947
[20]Norman T J,Grant C D,Magana D,et al.J.Phys.Chem.B,2002,106(28):7005-7012
[21]Shen M,Sun Y,Han Y,et al.Langmuir,2008,24(22):13161-13167
[22]Shen M,Chen W F,Yan C G.J.Phys.Chem.Solids,2007,68(12):2252-2261
Green Synthesis of Gold Nanoparticles and Deaggregating Effect of CTAB on Gold Nanoaggregates
DAI Jing-Tao1,3ZHU Jing2YANG Jin-Ming1,3SHEN Ming*,1,2
(1Key Construction Laboratory of Shoal Bioresource and Environment Protection of Jiangsu Province,Yancheng,Jiangsu 224002,China)(2College of Chemistry and Chemical Engineering,Yangzhou University,Yangzhou,Jiangsu 225002,China)(3School of Chemistry and Chemical Engineering,Yancheng Teachers University,Yancheng,Jiangsu 224002,China)
Gold nanoparticles were fabricated by a green method using tannic acid as a reducing and protecting agent at room temperature.The tannic acid-capped gold nanoparticles were characterized by UV-Vis,TEM,XRD,FTIR,respectively.The results show that the maximum absorption wavelength increases and the absorption band broadens,while the maximum absorbance decreases with the increase of the molar ratio of tannic acid to HAuCl4(nTA/nHAuCl4).TEM observation indicates that the increase in tannic acid amount could lead to the formation of flower-like gold nanoaggregates in the colloid systems.The experiments reveal that cetyltrimethylammonium bromide (CTAB)has a strong deaggregating effect on tannic acid-capped gold nanoaggregates.And CTAB can successfully deaggregate the gold nanoaggregates into monodispersed gold nanoparticles under vigorous stirring.In addition,the increase in the system temperature can obviously promote the deaggregating effect of CTAB on gold nanoaggregates.
tannic acid;gold nanoparticle;green synthesis;cetyltrimethylammonium bromide;deaggregating effect
O614.123;O648.16
:A
:1001-4861(2011)02-0308-07
2010-09-06。收修改稿日期:2010-10-27。
江苏省滩涂生物资源与环境保护重点建设实验室开放基金(No.JLCBE09003);国家自然科学基金(No.20773105)资助项目。
*通讯联系人。 E-mail:shenming@yzu.edu.cn,Tel:0514-87975590-9405;会员登记号:S06N7798M1006。
猜你喜欢
合成化学(2024年3期)2024-03-23 00:56:44
合成化学(2023年12期)2024-01-02 01:02:18
河南工业大学学报(自然科学版)(2021年6期)2022-01-26 06:36:08
装备制造技术(2020年4期)2020-12-25 05:25:52
中老年保健(2020年10期)2020-12-04 02:27:31
保健与生活(2020年10期)2020-05-28 13:47:03
健康博览(2016年11期)2016-12-23 21:03:38
无机盐工业(2016年4期)2016-03-15 18:34:14
应用化工(2014年9期)2014-08-10 14:05:08
石油化工(2014年1期)2014-06-07 05:57:08
