
关于化学反应表观活化能和指前因子的教学讨论
2011-09-26 01:22王新平王旭珍王新葵刘泽群牛家豪
大学化学 2011年3期
王新平 王旭珍 王新葵 刘泽群 牛家豪
(大连理工大学化学学院 辽宁大连 116024)
在世界范围内,人们已经发现并利用于工业过程的化学反应,90%以上是借助催化剂实现的[1]。而且,催化反应在工业反应中所占的比例,还在逐年增加,其中多相催化反应尤为重要。本科物理化学的反应动力学内容包括以下几个方面:反应速率与反应物浓度的关系(反应级数的确定),反应速率与温度的关系(表观活化能的测定),反应机理的提出及多相催化剂的基础知识等。基于上述催化反应过程的发展趋势,我们认为,关于反应动力学的本科生教学,应围绕催化过程和催化剂为中心展开,使学生掌握关于催化过程的化学反应动力学基础。
对于多相催化反应,当催化剂改变时,反应速率系数随之改变,但反应机理、反应活化能以及反应级数是否会改变,则与催化剂的活性中心结构是否改变直接相关。该内容是催化反应动力学创新思维的基础,但在以往的物理化学教材中却不够突出。经过对多年物理化学教学进行总结,我们认为,虽然在化学动力学中关于多相催化剂的基础知识涉及不多,但将其与Arrhenius方程中的指前因子和活化能相联系,则利于学生建立关于催化反应动力学创新思维,培养其创新能力。在此,我们从讨论Arrhenius方程表述的速率系数出发,给出这种相应关联性,以为教学同行参考。
1 Arrhenius方程与活化能的测定
对于速率有规律性地受反应温度影响的化学反应,Arrhenius提出的反应速率系数与温度之间关系的经验方程为:
(1)
该方程被称为Arrhenius方程,其中R为摩尔气体常量,k0和Ea是两个经验参量,分别称为指前参量和活化能。
化学反应的活化能通常在40~400kJ·mol-1之间[2]。活化能越高,表示反应对温度越敏感。因此,测定活化能数据,有利于优化反应条件,提高目的产物生成选择性。例如,反应物A和B可同时发生下列主、副反应:

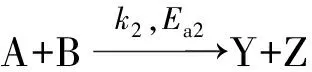
将式(1)定积分,得:
(2)
其中k(T2)和k(T1)分别为在温度T2和T1下的反应速率系数。对于上述两个平行竞争反应,由式(2)有:
(3)
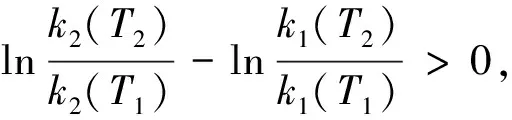
对于催化反应,获取活化能数据的另一个重要意义是,由活化能的相对高低可比较催化剂活性结构的优劣,从而为高活性催化剂的设计提供理论依据。催化剂活性的高与低,即催化剂在指定的反应条件下所给出的反应系数k的大与小。由Arrhenius方程可知,提高指前因子k0和降低反应活化能均可提高反应速率系数。但对用于多相反应的固体催化剂而言,二者指示的意义却是完全不同的(相应内容将在第3部分详述)。
关于获取活化能的实验方法,在理论上,只要能得到两个不同温度下的速率系数,根据式(2)即可算出反应的活化能。但是,为了避免出现较大的实验误差,实验室一般并不采用“两点法”,而是采用下述的“多点”直线法:
把式(1)两边取自然对数,得:
(4)
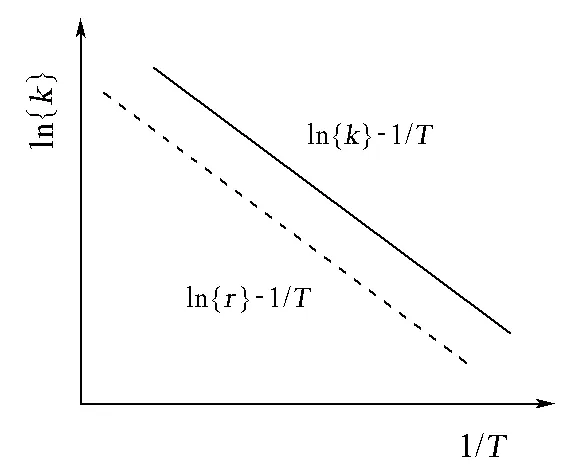
图1 符合Arrhenius方程的反应在不同反应温度下的直线和直线
由指定温度下的反应速率(r)计算反应速率系数,必须首先确定反应的级数,而后者又需要测定一系列反应时间下的反应物浓度后才能确定。因此,要想通过反应速率系数获得反应的活化能,便需要做很多研究工作。

ln{r}=ln{k}+αln{pA}+βln{pB}+…
(5)
将其代入式(4),有:
(6)

2 Arrhenius方程的指前因子与反应条件的关系
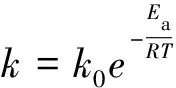


……(上述循环共重复n次。)

根据上述反应机理,有:

在直链反应过程中,由于中间物Cl和H的浓度不变,则用稳态法近似处理,有:
(7)
(8)
式中c(S)和c(M)分别表示系统中相应稳定质点和高能质点的点位(或吸收的光子)数。
由式(7),有:
k2c(Cl)c(H2)=k3c(H)c(Cl2)
代入式(8),有:
k1c(M)c(Cl2)=k4c(S)(c(Cl))2
则有:
于是得:
显然,对于该反应,k0的大小不仅与反应系统中高能量质点的浓度c(M)及稳定分子的浓度c(S)有关,还和链传递的活性质点Cl在链终止前所完成的链传递循环次数n的大小直接相关。在其他条件不变的情况下,n的大小受制于反应物的分压。这是因为,在一定条件下,反应物浓度的增大将增加链传递活性质点继续传递的概率,从而使n(平均值)增大。当然,n的大小还与稳定分子浓度c(S)的大小及反应器壁的表面积等诸多因素直接相关。
3 指前因子、活化能与催化剂物理化学结构的关系
对于气/固(或液/固)相催化反应,至少有一种反应物的分子被催化剂表面吸附活化,从而改变化学反应的途径、降低反应的活化能,使反应容易进行(图2)。

图2 反应的最低能量途径和相应活化能1 有催化剂;2 无催化剂
对于通常的催化剂而言,其微观表面并不均匀。在反应条件下,其表面上只有一些特殊的点位才吸附反应物分子并对其活化,从而起催化作用。这样的点位称为催化剂的表面活性位(或活性中心)。
催化剂活性的差异一般是由于其活性中心结构不同而导致对反应物活化能力的不同。改变催化剂的活性组分或加入电子型助催化剂,可使活性位的组成结构发生改变,从而提高活性中心对反应物的活化能力。由此所引起的反应速率系数的提高是由于进一步降低了反应的表观活化能。当然,此时活化能的降低可能来自下列两种情况之一:① 反应途径没有变化,表观活化能的降低仅是因速率控制步骤的活化能降低所致(图3A);② 改变了反应途径,从而降低了反应的表观活化能(图3B)。
上述两种情况经常在多相催化研究中出现。当催化剂的活性中心结构接近时(例如活性组分为同一族的过渡元素),反应活化能的改变完全可能是图3A的状况;而当催化剂的活性中心的结构相差较大时,则通常表现为图3B的状况。

图3 不同催化剂(1和2)上同一化学反应表观活化能不同的两种可能原因A 反应途径相同;B 反应途径不同
需要注意的是,催化剂活性不同(即反应速率系数不同),并不一定是由于其反应活化能不同。当催化剂活性中心的结构完全相同时,其反应活化能是相同的。在这种情况下,如果能够增加催化剂的比表面(1克催化剂所具有的表面积),或者提高催化剂单位表面的活性中心密度,都能有效增加单位质量催化剂的活性中心数目。这就相当于增加了催化剂用量。此时,反应速率系数的提高是由于Arrhenius方程中指前因子k0的提高。在制备催化剂时,通常总是希望得到大表面积的催化剂,其原因即在于此。使用大表面积的载体制备催化剂,其重要原因之一就是催化活性物能够更好地分散,以得到更多的活性中心,使Arrhenius方程中的指前因子k0较大。在制备催化剂时,使用结构型助剂(例如合成氨催化剂K2O-Fe-Al2O3中的Al2O3),则是为了抑制催化剂烧结(表面积迅速减小)导致的活性中心数目减少,即为了使k0保持不变。
具有更好活性中心结构的催化剂,其对应的反应活化能一定较小。虽然有时因其比表面较小或活性中心密度较小使得其给出的速率常数较小,但只要改进催化剂的制备方法,以提高催化剂的比表面及单位表面的活性中心密度,就能得到高活性的催化剂,从而有效提高反应的速率系数k。
参 考 文 献
[1] 美国国家催化委员会.催化展望.熊国兴,陈德安译.北京:北京大学出版社,1993
[2] 韩德刚,高执棣,高盘良.物理化学.北京:高等教育出版社,2001
[3] 吴越.催化化学(上册).北京:科学出版社,1998
猜你喜欢
河北果树(2021年4期)2021-12-02
上海公路(2019年3期)2019-11-25
福建基础教育研究(2019年10期)2019-05-28
石油石化绿色低碳(2019年6期)2019-02-13
中学化学(2017年5期)2017-07-07
浙江大学学报(工学版)(2016年11期)2016-06-05
中学化学(2016年4期)2016-05-30
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
中学化学(2014年1期)2014-04-23
