
CoB和CoB/SiO2对AP热分解的催化活性研究①
2011-09-26 03:10刘祥萱王煊军
固体火箭技术 2011年6期
李 茸,刘祥萱,王煊军
(西安高科技研究所503室,西安 710025)
0 引言
AP是固体推进剂的主要氧化剂之一,其热分解特性与固体推进剂的燃烧性能密切相关[1-4]。目前,关于过渡金属,其合金、氧化物、非晶合金及其他混合物作为AP基固体推进剂的燃速调节剂的研究很多,其中非晶态物质的催化性质较好,尤其是Co基非晶态合金[5-13]。采用化学还原方法制备的超细非晶态合金虽然表现出良好的催化性能,可有效提高催化活性和选择性,但由于其热稳定性很差,催化剂成本高,且与产物分离困难,工业化应用难度很大[14-18]。相对非负载催化剂,采用化学还原浸渍方法制备的负载型非晶态合金催化剂的热稳定性好,有效防止了催化剂有效成分的烧结,并在催化过程中更可保持其非晶态合金的性质,由于活性组分均匀分散在载体中,还原不仅能将活性组分在催化剂内均匀固定,而且保证了催化剂的高活性。采用负载法可通过选用不同的载体来任意调节非晶态合金催化剂的比表面积和孔结构,以适应不同反应类型对催化剂表面积和孔结构的要求,并增加催化剂的灵活性和应用领域,负载型非晶态合金催化剂是很有工业应用前景的催化剂[6,19-20]。
近年来,由于CoB合金的高机械硬度,强耐腐蚀性及极好的电、磁和催化特性,其越来越受关注[21-24]。但现有文献大多侧重于纳米级CoB和负载的CoB在生物、能源等方面的应用,而关于纳米CoB和负载的CoB合金对军用火炸药和火箭推进剂等方面的催化性能报道不多。
本文采用化学沉积方法制备纳米尺度的CoB和CoB/SiO2粉体,并通过热重-差热分析(TG-DTA)研究和比较CoB和CoB/SiO2对AP热分解的催化效果。
1 实验
1.1 催化剂的制备
以焙烧过的纳米SiO2作为载体(60 nm,500℃,5 h)及化学试剂KBH4和CoCl2·6H2O作为原材料,采用化学沉积方法制备CoB和CoB/SiO2催化剂。
将准确称取的SiO2(0.9 g)加入装有100 ml Co2+溶液(0.01 mol)体积为500 ml的圆底烧瓶中并封口,在室温下让SiO2载体在Co2+溶液中浸渍24 h。将圆底烧瓶置于冰水浴中,在快速搅拌下,将200 ml的KBH4溶液(0.02 mol,为防止水解,用 NaOH 溶液将KBH4溶液pH调至为12左右)在30 min内滴加至浸有载体的Co2+溶液中(Co2+与BH-4摩尔比为1∶2)。KBH4溶液与CoCl2·6H2O溶液一接触,反应立即进行,产生大量气泡,形成黑色乳液。均匀搅拌反应溶液至无气泡放出,反应结束。高速离心分离黑色乳液得黑色沉淀。反复用蒸馏水洗涤沉淀至pH=7,再用无水乙醇反复洗涤,所得湿粉体保存在EtOH中备用。湿粉体经低温(40℃,6~8 h)真空干燥,得CoB/SiO2干燥粉体催化剂。略去载体浸渍步骤,用同样工艺过程制备CoB催化剂。
1.2 催化剂的表征
催化剂的结构和物相在D/MAX-2400型X-ray衍射(XRD)仪(Cu-Kα辐射,工作电压 50 kV,工作电流150 mA,波长 λ =1.540 6 Å,扫描速度3(°)/min,扫描范围 20°~80°)、JEM-5600LV 型扫描电镜(SEM)和JEM-2000EM型高分辨率透射电镜(HR-TEM)上进行测试。催化剂的热力学稳定性在STA 449C型差扫描量热(DSC)仪上测试(N2气氛,N2流速20 ml/min,加热速率20℃/min)。
1.3 催化活性测试
将催化剂(0.02 g)与尺度为 16 μm 的 AP(0.98 g)以质量比1∶49在玛瑙研钵中混合,并加入1 ml无水乙醇研磨1 min。混合物在70℃真空干燥1 h。用TGA/SDTA851E/LF/1600型热重-差热分析(TG-DTA)仪测试混合物(N2气氛,N2流速50 ml/min,加热速率10℃/min)。
2 结果与分析
2.1 CoB和CoB/SiO2的物相
图1是CoB和CoB/SiO2催化剂的XRD图谱。2个图谱都不存在尖锐的衍射峰,均在2θ=40°~50°左右出现了强度较小的“馒头”状弥散衍射峰。这一结果与非晶态结构CoB的衍射图谱一致[6],说明载体SiO2表面负载上了CoB粒子,且CoB和CoB/SiO2都是非晶态结构。另外,弥散峰的宽化说明CoB粒子在SiO2载体表面的分散性较好,而衍射峰的强度较小,说明制备的CoB和CoB/SiO2催化剂粉体颗粒尺度很小。

2.2 CoB和CoB/SiO2的结构和形貌
在透射电镜和扫描电镜上测试CoB和CoB/SiO2催化剂的物相。图2为2种催化剂的纳米尺度SEM照片。从照片看,CoB团聚严重,表现出不规则的网络状微观结构,网状结构存在大量层叠的间隙和纳米级孔洞。SiO2载体呈现较透明的絮状准颗粒结构和明显团聚状态。CoB/SiO2也表现出类似的团聚现象和不规则絮状准颗粒结构,粉体颗粒尺度和形貌都近似于纳米SiO2载体。
图3是2种催化剂不同放大倍数的TEM照片及选区电子衍射(SAED)照片。照片说明,在100 nm尺度下,CoB是由交错的网状物质与镶嵌在其中的大量颗粒物所组成的团聚体。团聚体的尺度为100 nm左右,但在10 nm尺度下,可看见团聚体中分散有大量尺度为5 nm左右甚至小于5 nm的颗粒,这些颗粒应是纳米晶体粒子。这些尺度极小且活性很高的纳米晶体粒子极不稳定,相互团聚粘连形成CoB的特殊网状结构。由于纳米SiO2载体的不规则形状,在50 nm及10 nm尺度下,可清楚地看到絮状的SiO2载体表面沉积了大量的CoB粒子,且沉积粒子分布较均匀。10 nm尺度下,CoB/SiO2结构中也有大量5 nm左右的晶体粒子。CoB和CoB/SiO2的选区电子衍射照片都表现为非晶结构所特有的衍射晕圈而不是清晰的点,且晕圈中都有很多边界模糊的弥散的衍射环,说明CoB和CoB/SiO2整体都表现出非晶体相的性质,并验证了二者结构中纳米晶的存在。SAED结果与XRD结果一致,证明了CoB和CoB/SiO2都具有典型的短程有序、长程无序的非晶态结构特征。2种催化剂的结构中大量存在5 nm左右的纳米晶体粒子,CoB结构中大量层叠的间隙和纳米级孔洞,以及CoB/SiO2近似于纳米SiO2载体的尺度和形貌,都有利于增加催化剂的活性位置。

2.3 CoB和CoB/SiO2的催化活性
为便于比较,对纯AP及AP与SiO2、CoB和CoB/SiO2的混合样做热重-差热分析(SiO2、CoB和 CoB/SiO2的质量分数均为5%),TG-DTA曲线见图4,对应的TG-DTA数据列于表1。表1中,Tσ为相转变温度;Tp为热分解峰温;Te为热分解起始温度;Tt为热分解终止温度;MR为质量残留。TG-DTA曲线显示,纯AP在250℃存在1个吸热峰,对应由斜方晶型向立方晶型转变的固-固相转变过程,在311.80℃和450.14℃存在的2个放热峰,分别对应低温分解和高温分解2个放热阶段。这2个分解放热阶段与TG曲线上的2段失重过程相对应,339.58℃左右低温分解过程结束,整体分解反应至468.41℃完全结束。
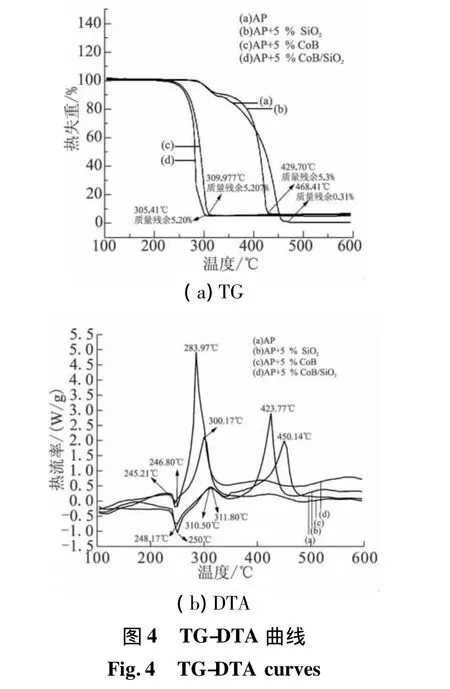
图4中,DTA曲线(b)~(d)的吸热峰温(Tσ)均在250℃附近对应纯AP吸热峰,说明空白SiO2、CoB和CoB/SiO2的加入并没有改变AP晶相转变峰的位置。但对照图4曲线和表1可看出,空白SiO2、CoB和CoB/SiO2均促进了AP的热分解,表现为DTA曲线上AP的高温分解峰温(Tp)大幅度下降,高温与低温热分解峰之间的温度区间变窄,整体分解反应的终止温度(Tt)提前。
加入5%的SiO2,SiO2/AP混合样的DTA曲线上低温Tp无明显改变,但高温Tp及Tt分别为423.77℃和429.70℃,较纯AP均提前了27℃左右。因此,纳米SiO2载体对AP的高温分解有较明显催化活性。
分别加入 5%的 CoB和 CoB/SiO2,CoB/AP和CoB/SiO2/AP混合样的DTA曲线上高温分解峰均提前,与低温分解峰叠加只表现一个低温热分解阶段,TG曲线上无拖尾峰。CoB/AP的低温Tp=300.17℃,较纯AP的低温和高温Tp分别提前了11.63℃和150℃。CoB/SiO2/AP的低温 Tp=283.97℃,较 CoB/AP的低温Tp提前16.2℃,较纯AP的低温和高温Tp分别提前了 27.83℃和 166.2℃。CoB/AP和 CoB/SiO2/AP混合样的反应Tt分别为310℃和305.41℃,较纯AP整体反应Tt(468.41℃)分别提前158.41℃和163℃。纯AP分解结束质量残余(MR)为0.31%,而CoB/AP和CoB/SiO2/AP混合样分解结束MR分别为5.21%和5.20%,说明CoB和 CoB/SiO2的加入量基本没变。因此,CoB和CoB/SiO2都对AP热分解表现出催化性质,尤其对AP高温热分解阶段有明显催化活性。CoB/SiO2的催化效果更为突出。
2.4 CoB和CoB/SiO2的热稳定性
非晶态合金是一种亚稳定状态,加热过程中向较稳定的非晶态或晶态转变,放出热量,在DSC曲线上表现为放热峰。图5是制备的CoB和CoB/SiO2催化剂的DSC曲线。CoB和CoB/SiO2的DSC曲线都有1个吸热峰和多个放热峰。2个DSC曲线在101.2℃附近出现的吸热峰,对应于吸附水的脱附。继续升温,CoB的DSC基线向放热方向漂移,表明CoB合金微粒的结构在逐渐变化。在268.7℃和346.2℃出现的弱放热峰,归于CoB非晶态合金从无序堆积向有序堆积的转变过程,也意味着非晶态合金开始发生晶化[14]。在474.2℃出现的强放热峰及在529.1℃和687.4℃出现的较强放热峰,归因于CoB非晶态合金的多步晶化过程。
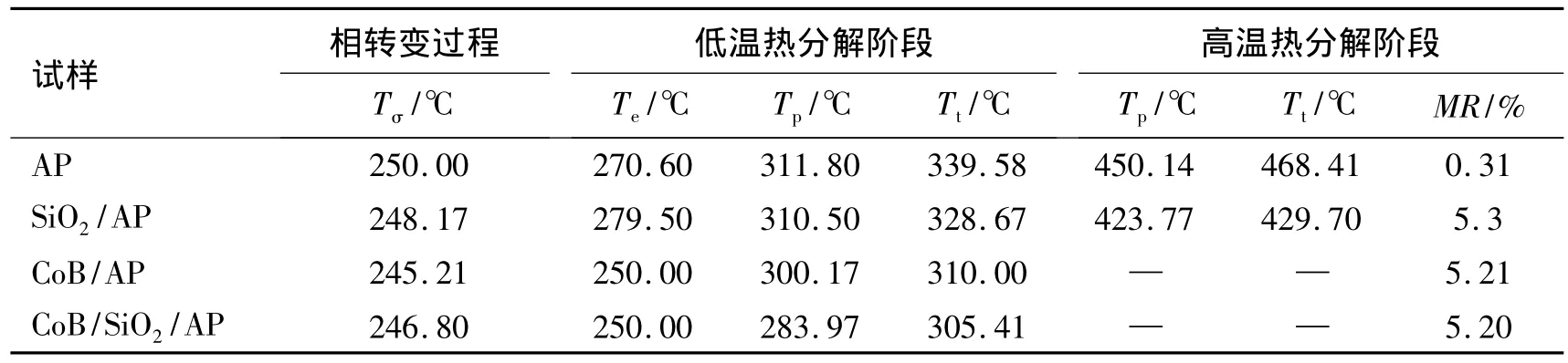
表1 TG-DTA数据Table 1 TG-DTA data

CoB/SiO2的DSC曲线直到500℃之前,曲线都处于一个连续、平稳、但较弱的放热过程,归因于微粒结构从无序到有序的逐渐转变过程。虽然500~800℃间DSC 曲线漂移至吸热过程,但在 587.3、650.7、697.7℃出现了朝向放热方向的峰,这3个峰归因于CoB非晶态合金的晶化。CoB/SiO2的DSC曲线向吸热方向的大幅漂移,可能是纳米CoB粒子分散在纳米级的SiO2载体中抑制了CoB粒子的晶化引起的。比较CoB/SiO2和CoB的热稳定性发现,前者在500℃之前基本是热稳定的,而后者在268.7℃已开始缓慢晶化;进行多步晶化时,前者的晶化温度比后者分别推迟了113.1、121.6、10.3 ℃左右。结果说明,纳米 SiO2载体的引入,延缓了热处理过程中CoB非晶态合金的彻底晶化,提高了CoB粉体的热稳定性。据文献[15]报道,在非晶态合金晶化过程中,非晶态颗粒的团聚是晶化的前提。因此,认为纳米SiO2载体能提高CoB催化剂的热稳定性,主要原因是CoB合金在比表面积很大的纳米SiO2载体上得到了较好的分散。载体与非晶态合金之间存在的物理作用(如空间结构束缚或静电作用)或化学作用(如化学键),使得非晶态合金纳米颗粒之间不易互相团聚,从而抑制了合金组分晶粒的析出,提高了CoB非晶态合金的晶化温度。CoB/SiO2粉体较好的热稳定性更有利于其在AP的催化热分解反应中发挥催化活性。
2.5 催化机理分析
AP的热分解分为低温热分解阶段(300~330℃)和高温热分解阶段[25](450~480℃)。人们普遍认为AP的低温热分解阶段是固-气多相反应,初始分解是质子从NH4+转移到ClO4-生成NH3和HClO4的离解过程,存在着升华与分解2个竞争过程。首先是离解与升华:
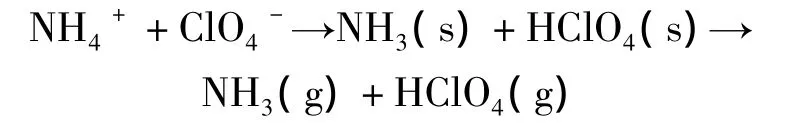
低温分解阶段主要的热分解反应是HClO4的降解及HClO4的分解产物氧化NH3的氧化反应。此阶段,主要中间产物HClO4除自身降解消耗一部分外,存在一个积累过程,因此AP粒子表面吸附的NH3不能全部由HClO4的分解产物所氧化,当NH3覆盖在AP粒子表面和催化剂粒子表面的全部活化中心(反应中心)时分解过程停止,催化剂对AP低温阶段的反应活性表现的不突出。在高温条件下,由于NH3的解吸,NH3被HClO4降解产物氧化的反应及HClO4、NH3与其他中间分解产物的反应重新开始并加剧。此阶段,气相反应是高温反应的控制步骤。因此,催化剂对AP高温阶段的分解表现出优异的催化效率[26-27]。
本文制备的CoB和CoB/SiO2显著降低AP的高温热分解温度。据此可认为,CoB和CoB/SiO2增大了HClO4的反应速率,从而促进了AP的高温分解历程。根据本文实验数据和其他研究结果,CoB和CoB/SiO2的结构与形貌及过渡金属Co活泼的化学性质是催化剂优异催化作用的可能原因。催化剂的热力学稳定性对其催化效果也有影响。
根据价键理论[28],过渡金属电子层结构的s带和d带之间有交迭,因而影响d电子的填充程度,d带出现了空穴,即“d带空穴”。从催化反应的角度看,d带空穴的存在,使金属有从外界接受电子和吸附物种,并与之成键的能力。金属 Co的基态电子层结构是Ar3d74s2,d带有1.6个空穴。在 AP的高温分解阶段,Co通过大量吸附NH3、HNO和O2等气体中间产物削弱N—H,H—O及Cl—O化学键,从而加速NH3的氧化反应及HNO、HClO4、NH3和其他中间产物间的相互反应,并最终显著降低AP的高温热分解温度。CoB网状结构中大量的交叉点与间隙,CoB/SiO2的纳米SiO2载体及2种催化剂结构中,大量存在的5 nm左右的纳米晶体粒子则为催化剂提供大量活性位置去吸附气体中间产物。介于SiO2载体对AP微弱的催化效果,添加量同为5%,CoB/SiO2的催化活性高于CoB,归因于其较高的热稳定性。在AP的高温分解温度范围内,CoB/SiO2没有发生明显结晶过程。因此,其活性点不会因为结构的改变而减少,而CoB在此温度范围是不稳定的。
3 结论
(1)采用化学沉积方法分别制备了纳米尺度CoB和SiO2负载的CoB/SiO2催化剂,2种催化剂均表现出非晶体特征,CoB有不规则网状结构。
(2)质量分数5%的CoB和CoB/SiO2可将AP高温热分解峰的温度分别降低150、166.2℃。CoB和CoB/SiO2对AP热分解都表现出催化活性,CoB/SiO2的催化活性更强。
(3)负载型CoB/SiO2多步晶化的放热峰比CoB分别推迟了 113.1、121.6、10.3 ℃ 左右,CoB/SiO2较高的热力学稳定性更有利于其催化活性的履行。
[1]Leu A L,Yeh T F,Chang F M.Burning behavior of composite solid propellant containing porous ammonium perchlorate[J].Propellants,Explos.,Pyrotech.,1989,14:108-112.
[2]Aleksandr V D.Physicochemical analysis of explosions due to impact in mixed paste propellants[J].Propellants,Explos.,Pyrotech.,2005,30:244-249.
[3]Baptiste L,Philippe G.Experimental investigation on the heterogeneous kinetic process of the low thermal decomposition of ammonium perchlorate particles[J].Propellants,Explos.,Pyrotech.,2009,34:59-71.
[4]Said A A,Al Qasmi R.The role of copper cobaltite spinel,CuxCo3-xO4during the thermal decomposition of ammonium perchlorate[J].Thermochim.Acta,1996,275:83-91.
[5]Liu H B,Jiao Q Z,Zhao Y.Mixed oxides derived from Cu-Co layered double hydroxide nanorods:Preparation,characterization and their catalytic activities[J].J.Alloys Compd.,2010,496:317-323.
[6]Li H X,Li H,Zhang J.Ultrasound-assisted preparation of a highly active and selective Co-B amorphous alloy catalyst in uniform spherical nanoparticles[J].J.Catal.,2007,246:301-307.
[7]Ping C,Li F S,Jian Z.Preparation of Cu/CNT composite particles and catalytic performance on thermal decomposition of ammonium perchlorate[J].Propellants,Explos.,Pyrotech,.2006,31:452-455.
[8]Yu Z X,Chen L F,Lu L D.DSC/TG-MS study on in situ catalytic thermal decomposition of ammonium perchlorate over CoC2O4[J].Chinese Journal of Catalysis,2009,30:19-23.
[9]Xu H,Wang X B,Zhang L Z.Selective preparation of nanorods and micro-octahedrons of Fe2O3and their catalytic performances for thermal decomposition of ammonium perchlorate[J].Powder Technol.,2008,185:176-80.
[10]Duan H Z,Lin X Y,Liu G Q.Synthesis of Co nanoparticles and their catalytic effect on the decomposition of ammonium perchlorate[J].Chinese Journal of Chemical Engineering,2008,16(2):325-328.
[11]Chen L J,Li L Q,Li G S.Synthesis of CuO nanorods and their catalytic activity in the thermal decomposition of ammonium perchlorate[J].Journal of Alloys and Comp.,2008,464:532-536.
[12]Li R,Liu X X,Wang X J.Synthesis of Ni,Ni-P and Ni-B nanoparticles and their catalytic effect on the decomposition of ammonium perchlorate[J].Journal of Solid Rocket Technology,2008,6:295-298.
[13]Singh Kapoor I P,Srivastava P,Singh G.Nanocrystalline transition metal oxides as catalysts in the thermal decomposition of ammonium perchlorate[J].Propellants,Explosives,Pyrotechnics,2009,34(4):351-356.
[14]闫世润,乔明华,范康年.非晶态合金催化剂的研究进展[J].石油化工,2007,36(3):213-221.
[15]左满宏,王志,刘恩利.非晶态合金催化剂的表征与应用[J].广州化工,2007,35(1):5-9.
[16]谷燕,刘贵昌.化学镀在制备纳米材料中的应用[J].材料保护,2006(1):40-43.
[17]李茸,刘祥萱,王煊军,等.纳米金属催化剂的制备及其后处理[J].新技术新工艺,2007,237:86-89.
[18]马延凤,张明慧,李伟,等.高比表面积NiP非晶态合金的制备及其催化性能[J].催化学报,2004,25(12):973-978.
[19]Li C,Ma Z Y,Zhang L X.Preparation of Ni/TiO2nanoparticles and their catalytic performance on the thermal decomposition of ammonium perchlorate[J].Chinese Journal of Chemistry,2009,27(10):1863-1867.
[20]Li H X,Wang W J,Li H.Crystallization deactivation of Ni-P/SiO2amorphous catalystand the stabilizing effect of silica support on the Ni-P amorphous structure[J].Journal of Catalysis,2000,194:211-221.
[21]Bae J W,Kim S M,Kang S H.Effect of support and cobalt precursors on the activity of Co/AlPO4catalysts in Fischer-Tropsch synthesis[J].J.Mol.Catal.A:Chem.,2009,311:7-16.
[22]Liaw B J,Chen C H,Chen Y Z.Hydrogenation of fructose over amorphous nano-catalysts of CoNiB and polymer-stabilized CoNiB[J].Chem.Eng.J.,2010,157:140-145.
[23]Xu J K,Zhou W,Li Z J.Biogas reforming for hydrogen production over nickel and cobalt bimetallic catalysts[J].Int.J.Hydrogen Energy,2009,34:6646-6654.
[24]Buan N R,Metcalf W W.Methanogenesis by methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase[J].Mol.Microbiol,2010,75:843-853.
[25]Boldyev V V.Thermal decomposition of ammonium perchlorate[J].Thermochim.Acta,2006,443:1-36.
[26]Rosser W A,Inami S H.Thermal decomposition of ammonium perchlorate[J].Combust.Flame,1968,12:427-435.
[27]Pellet G L,Cofer W R.Thermal decomposition of ammonium perchlorate by rapid heating,seventh AIAA aerospace science meeting[C].New York,1969:141.
[28]Bamford C H,Tipper C F H.Decomposition reactions of solids[M]//Comprehensive Chemical Kinetics,Amsterdam,1980,22:115-246.
猜你喜欢
铝加工(2022年3期)2022-11-24
辽宁化工(2022年8期)2022-08-27
材料与冶金学报(2022年2期)2022-08-10
——水热过程影响机制
化工进展(2021年11期)2021-11-30
石油炼制与化工(2021年9期)2021-09-15
建材发展导向(2021年13期)2021-07-28
粉末冶金技术(2021年3期)2021-07-28
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
分析化学(2018年1期)2018-01-18
