
反相离子对高效液相色谱法测定硫酸奈替米星滴眼液含量
2011-09-19 08:46杨帆
亚太传统医药 2011年12期
杨 帆
(广西医科大学附属第四医院/柳州市工人医院 药剂科,广西 柳州545001)
奈替米星(Netilmicin)是一种新的氨基糖苷类药物,为半合成的氨基糖苷类抗生素,对革兰氏阴性菌和部分革兰氏阳性菌具有抑菌和杀菌作用,不良反应较同类药物小,国内外已广泛应用于临床。将其制成滴眼液可用于多种细菌及耐药菌造成的眼部感染。该类抗生素多为紫外末端吸收或无紫外吸收,故很难用紫外分光光度法直接测定其含量。中国药典2005版采用抗生素微生物检定法测定硫酸奈替米星含量,但该法操作繁琐、费时。近年来也有文献报道采用旋光法、柱前衍生化法、高效液相色谱-蒸发光散射检测法[4]等,前两种方法受实验影响因素较多,后者因需对检测结果进行数学处理而稍显复杂,我们建立了反相离子对高效液相色谱法直接测定其含量。通过在pH=3.0的条件下,在1.5%的磷酸二氢钾溶液中加入离子对试剂十二烷基硫酸钠,被测物奈替米星与十二烷基硫酸钠生成离子对,并用乙腈调节流动相的极性,以调节出峰时间,主峰与杂质峰分离良好。此法对硫酸奈替米星滴眼液的含量测定,取得了较满意的结果,现报道如下。
1 仪器、试药及实验条件
1.1 仪器与试药
LC-10ATvp高效液相色谱仪,包括SPD-10Avp紫外检测器、CLASS-VP Ver 6.12SP1工作站(日本岛津公司);UV-2401-PC紫外分光光度计(日本岛津公司);电子PHS-3F酸度计(上海电子光学技术研究所);BP211D电子天平(德国赛多利斯公司);KDC-16H高速台式离心机(科大创新股份有限公司中佳分公司);0412-1离心机(上海手术器械厂)。注射用硫酸奈替米星(南京圣和药业有限公司,批号:200608232青岛第三制药厂,批号070801);硫酸奈替米星标准品(中国药品生物制品检定所,批号:0355-200001);乙腈、甲醇为色谱纯,其他试剂为分析纯。
1.2 实验条件
(1)以1.5%磷酸二氢钾溶液(用磷酸调pH为3.0,十二烷基硫酸钠10mmol/L)-乙腈(85:15)为流动相。实验色谱柱:Diamonsil C18柱(250mm×4.6mm,5μm),流速:0.8mL/min;柱温:30℃;进样量:20μL;检测波长:205nm。
(2)含量测定按照高效液相色谱法《中国药典2005年版》二部附录V附录VD测定。色谱条件与系统适用性实验用十八烷基硅烷键合硅胶为填充剂;以1.5%磷酸二氢钾溶液(用磷酸调pH 为3.0,十二烷基硫酸钠10mmol/l)-乙腈(85:15)为流动相;检测波长为205nm。理论塔板数按硫酸奈替米星计算不低于1 500,硫酸奈替米星峰与各杂质峰之间能良好地分离。
2 系统适用性实验
按本品种项下要求对仪器进行系统适用性实验,应能达到规定的要求,方能进行实验。按该色谱系统分析条件下色谱峰的理论塔板数、分离度、重复性应达到要求。
色谱柱的理论塔板数(n)在所选择的条件下,注入供试品溶液,记录色谱图,量出供试品主成分色谱峰的保留时间tR和半峰高宽(Wh/2)按n=5.54(tR/Wh/2)2计算色谱柱的理论塔板数,经计算所得的理论塔板数符合要求。
分离度。定量分析时,为便于准确测量,要求定量峰与其他峰之间有较好的分离度,分离度(R)的计算公式为:R=2(tR1-tR2)/(W1+W2),式中tR2为相邻两峰中后一峰的保留时间;tR1为相邻两峰中前一峰的保留时间,W1和 W2为此相邻两峰的峰宽;根据实验得出的分离度大于1.5,符合要求。
重复性。取该品种项下的标准溶液,连续进样6次,峰面积测量值的相对标准偏差为0.58%,实验证明重复性符合要求。
3 方法与结果
3.1 滴眼液的处方
硫酸奈替米星5.0g,硼酸10.5g,硼砂2.87g,羟苯乙脂0.3g,注射用水加至1 000mL。
3.2 滴眼液的配制
取羟苯乙脂加适量新鲜注射用水溶解后,依次加入硫酸奈替米星、硼酸、硼砂使其全部溶解,滤过,自滤器上加注射用水至全量,搅匀,除菌,分装即得。
3.3 滴眼液含量测定
3.3.1 专属性实验(辅料干扰实验)
取处方量的辅料及注射用水按处方比例配制成辅料溶液,精密吸取2mL,加流动相稀释至25mL容量瓶,摇匀,过0.45μm滤膜,取20μL注入高效液相色谱仪,记录色谱图。结果表明辅料在此条件下无吸收,对奈替米星的含量测定无干扰。

图1 标准品色谱

图2 供试品色谱

图3 空白辅料实验色谱
3.3.2 标准曲线的绘制
精密称取硫酸奈替米星标准品适量(相当于125mg),用流动相配成5.0mg.mL-1的溶液,精密量取此溶液用流动相稀释成0.1、0.2、0.4、0.6、0.8、1.0mg.mL-1的溶液,摇匀,过0.45μm滤膜,分别取20μL进样测得峰面积。以浓度为横坐标,峰面积为纵坐标回归的直线方程为Y=3.129×106X+15 029,r=0.999 8,n=6。结果表明硫酸奈替米星在0.1~1.0mg·mL-1的浓度范围内,与峰面积线性关系良好。标准品重124.8mg,初体积25mL,初浓度4.992。
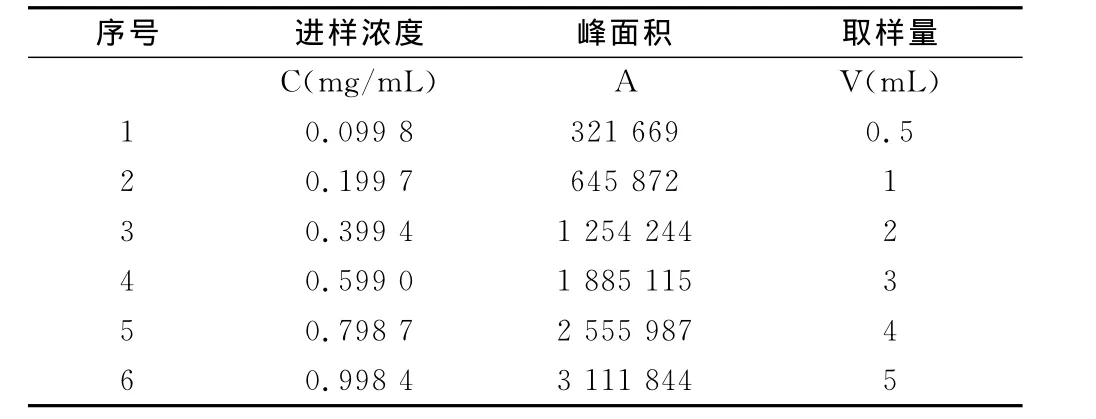
表1 硫酸奈替米星标准曲线的制备

图4 标准曲线
3.3.3 稳定性试验
取标准曲线下浓度为0.40mg·mL-1的溶液分别在0h,1h,2h,3h,4h,8h,12h,24h测定,奈替米星在该流动相中24h内是稳定的,RSD为0.84%,n=8。

表2 硫酸奈替米星稳定性考察 (n=8)
3.3.4 精密度试验
取标准曲线项下浓度为0.40mg.mL-1的溶液,重复进样6次,每次20μL,测定奈替米星的峰面积,结果RSD为0.58%。(见表3)

表3 硫酸奈替米星精密度试验 (n=6)
3.3.5 重复性试验
取同一批样品,按“样品测定”项下的方法测定6次,其含量测定结果的RSD为0.92%。见表4。

表4 硫酸奈替米星滴眼液含量测定重复性试验(n=6)
3.3.6 加样回收率试验
按处方量分别配制高(120%)、中(100%)、低(80%)的硫酸奈替米星滴眼液,取样2mL用流动相溶解25mL容量瓶定容后,过0.45μm滤膜,各取20μL进样,测定奈替米星峰面积并计算回收率,测得平均回收率为100.1%,RSD为1.16%,n=9,结果见表5。

表5 硫酸奈替米星滴眼液含量测定回收率实验
3.3.7 样品含量测定
取硫酸奈替米星滴眼液2mL,用流动相稀释后配成0.4mg·mL-1的溶液;另取硫酸奈替米星标准品适量,用流动相配成0.4mg.mL-1的溶液。分别取上述溶液20μL进样,按外标法以峰面积计算含量。三批样品测定结果分别为100.9%、98.1%和99.7%。

表6 硫酸奈替米星滴眼液的含量测定
4 讨论
(1)测定波长的选择:取硫酸奈替米星标准品适量,用流动相溶解后,以流动相为空白,在190~400nm波长范围内扫描,硫酸奈替米星在205nm波长处有较大吸收峰,选择205nm波长为测定波长。同时选用较低背景吸收的乙腈作为流动相中的有机相时,基线稳定,对测定无干扰。
(2)色谱柱的选择:反相离子对色谱法要求尽可能选择表面覆盖度高且疏水性强的键合相,如C8或C18键合相。长链烷基可使溶质的k增大,分离选择性改善,使载样量提高,长链烷基键合相的稳定性也更好。因此,本实验选择了以十八烷基硅烷键合硅胶为填充剂的Diamonsil C18柱为分析色谱柱。且本实验条件流动相的pH值较低,而Diamonsil C18柱的pH适用范围也较广(1.5~8.0),故该柱符合实验要求并能得到较好的分离效果。
(3)流动相的选择:反相键合相色谱法中,流动相一般以极性最强的水为基础溶剂,加入甲醇、乙腈等极性调节剂。由于在检测波长205nm处甲醇的吸光度较乙腈大许多,检测时产生的噪音过大,而且甲醇的黏度也比乙腈大,会产生过高的柱压,故选择乙腈作为调节剂。乙腈比例越大,流动相的洗脱能力越强,奈替米星的容量因子就越小,保留时间也越短,直接影响到主峰与杂质的分离效果。本实验选取1.5%磷酸二氢钾溶液(用磷酸调pH为3.0)-乙腈(85:15)为流动相,并用此流动相作为溶媒溶解样品使得用高效液相色谱法直接测定硫酸奈替米星含量成为可能,且辅料在此条件下无吸收,样品在此流动相中稳定性良好。
(4)氨基糖苷类抗生素多为紫外末端吸收或无紫外吸收,该类抗生素用HPLC法进行含量测定时多采用柱前衍生化法。但衍生化反应影响因素多,增加了方法的系统误差而且操作繁琐。本文建立了奈替米星质量控制的有效方法。
[1]陈新谦.新编药物学[M].第15版.北京:人民卫生出版社,2003:79.
[3]王建,胡小君,倪坤仪.HPLC-ELSD和 HPLC-MSn分析硫酸奈替米星及其注射液[J].中国药学杂志,2006,41(16):1268-1271.
[4]韦敏,梁建成,肖亿.改进微生物法监测人体内硫酸奈替米星血药浓度[J].2005,14(2):126-127.
[5]杨毓芬.反相离子对色谱法检测3种氨基糖苷类抗生素的混合样品[J].中国抗生素杂质,1997,22(2):152-153.
猜你喜欢
中国典型病例大全(2022年13期)2022-05-10
父母必读(2021年10期)2021-10-25
新疆钢铁(2021年1期)2021-10-14
中国食品(2021年18期)2021-09-28
家庭医药(2019年3期)2019-03-31
科技资讯(2018年16期)2018-10-26
理科考试研究·高中(2017年7期)2017-11-04
科技视界(2016年19期)2017-05-18
中学生理科应试(2017年2期)2017-04-01
润滑油(2015年4期)2015-11-20
