
球磨EMD对熔融浸渍法合成锰酸锂的影响
2011-09-18 06:20:32刘宏基刘兴泉向小春
电池 2011年6期
刘宏基,刘兴泉,王 超,向小春
(电子科技大学微电子与固体电子学院,电子薄膜与集成器件国家重点实验室,四川成都 610054)
高温固相法[1]是LiMn2O4的传统合成方法,流程简单,适于大规模生产,但需要长时间的高温处理,且产物颗粒较大、物相分布不均匀,电化学性能欠佳。作为对高温固相法的改进,M.Yoshio等[2]以熔融浸渍法合成了锂锰氧化物。该方法利用LiOH、LiNO3等锂盐熔点较低、锰的氧化物大多具备高熔点、呈多孔结构的特性,将锂盐与锰源混合后,在较低的温度下保持一段时间,使锂盐分解浸渍到锰盐的孔洞中,再在较高的温度下热处理,获得目标产物。Y.Xia等[3]以熔融浸渍法合成LiMn2O4,分析了产物的形貌、结构、电化学性能及高温稳定性,证实熔融浸渍法合成的LiMn2O4可兼具较高的比容量和优良的循环稳定性。与高温固相法相比,熔融浸渍法在一定程度上增加了分子接触面积,使发生反应所需的温度降低,且能使生成物的粒度分布更均匀[4]。
本文作者以颗粒尺寸较大的工业电解二氧化锰(EM D)为锰源,以熔融浸渍的处理方式合成LiMn2O4材料。锰源的粒径较大,直接与锂源混料烧结会阻碍材料中离子的扩散,因此在混料前先对锰源进行球磨处理。球磨将赋予材料大量的机械能,可降低锰源粒径,增大比表面积和表面能,缩短反应中离子的固相扩散距离,增大反应活性;但长时间的球磨可能破坏锰源原有的多孔结构,阻碍锂盐的熔融浸渍过程,因此,锰源的球磨时间可能存在一段最佳值。为此,对球磨时间进行了分析;同时,对比了人工研磨锰源与机械球磨锰源粒径、形貌的差异,以及对最终产物性能的影响。
1 实验
1.1 LiMn2O4的熔融浸渍法合成
以LiOH·H2O(成都产,95%)为锂源,EMD(湘潭产,工业级,94%)为锰源,以熔融浸渍法合成尖晶石 LiMn2O4。EMD在混料前先进行人工研磨或机械球磨,锰源处理方式与样品编号的关系见表1。
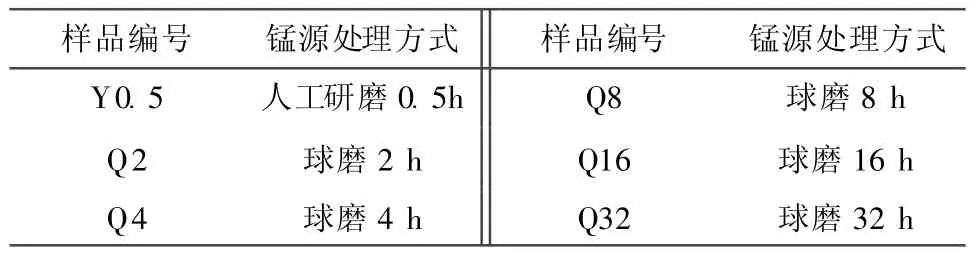
表1 各LiMn2O4样品对应的锰源处理方式Table 1 Manganese source treating method corresponding to each LiMn2O4samples
在QM-3SP2型行星式球磨机(南京产)上进行机械球磨,球料比为8∶1,转速为200 r/min;在玛瑙碾钵中进行人工研磨,时间为0.5 h。按n(Li)∶n(Mn)=1.05∶2.00混合锂源与处理后的锰源,再转移至刚玉坩埚中,在电阻炉中、空气气氛下进行热处理:先以5℃/min的速度升温至470℃并保温6 h,冷却后稍微研磨,使预烧后的粉体散开,再将预烧料以5℃/min的速度升温至750℃并保温24 h,随炉冷却后,得到样品。
1.2 LiMn2O4的形貌及结构分析
在6490LV型扫描电子显微镜(日本产)上对EMD及LiMn2O4样品进行形貌分析;在Pro MPD DY1291型粉末衍射仪(日本产)上对LiMn2O4样品进行结构分析。
1.3 模拟电池的装配
将合成的LiMn2O4样品与导电剂乙炔黑(四川产,工业级)、粘接剂聚偏氟乙烯(四川产,工业级)按质量比 85∶10∶5混匀,涂覆在20μ m厚的铝箔(成都产,99.5%)上,制成正极基片,最后在120℃下真空(真空度为-0.1 MPa)干燥8 h,制成φ=10 mm的正极片,其中活性物质含量为85%。以Celgard 2400(美国产)为隔膜,1.0 mol/L LiPF6/EC+DEC+DMC(体积比1∶1∶1,韩国产)为电解液,金属锂片(成都产,99.5%)为负极,在氩气气氛手套箱中装配内径为 12 mm、外径为20 mm、高度为20 mm的自制模拟电池。
1.4 电化学性能测试
装配的电池在DC-5型电池测试仪(上海产)上以0.5C(60 mA/g)的电流进行一次恒流预充放电(3.40~4.35 V),进行激活,再进行测试。在Corrtest CS350型电化学工作站(武汉产)上进行循环伏安测试,扫描速率为0.15 mV/s,电压为3.4~4.5 V;在DC-5型电池测试仪上进行充放电测试,以0.5C在3.40~4.35 V恒流充放电20次。
2 结果与讨论
2.1 SEM分析
图1为经不同方法处理的EMD的SEM图。

图1 经不同方法处理的EMD的SEM图Fig.1 SEM photographs of EMD treated by different methods
从图1a可见,未经处理的EMD呈片状,颗粒形状不规则,大部分颗粒的尺寸超过20 μ m;从图1b可见,人工研磨后的EMD,颗粒尺寸有所下降,但粒径差异较大,部分大颗粒的尺寸仍在 5μ m以上。相比人工研磨的EMD,球磨2 h的EMD(图1c)颗粒尺寸趋于均匀,整体粒径降至 1~2 μ m,层状结构保持完好,可看到层状结构之间的孔洞;随着球磨时间的延长,EMD颗粒尺寸持续下降,球磨4 h的EMD(图1d)粒径更均匀;球磨8 h的EMD(图 1e)中已看不到大于 1 μ m的微粒,且EM D颗粒之间仍保持较明显的层状结构;球磨16 h的EMD(图1f)颗粒细小且均匀,但层状结构大部分被破坏;球磨32 h的EMD(图1g)小颗粒出现了明显的团聚,原本的多孔结构也被完全破坏。从整体上来看,球磨后的EMD比人工研磨的,具有更小的粒径和更均匀的粒度分布,随着球磨时间的延长,在颗粒尺寸逐渐下降的同时,原有的层状结构也逐步被破坏。
图2为合成的LiMn2O4样品的SEM图。

图2 合成的LiMn2O4样品的SEM图Fig.2 SEM photographs of prepared LiMn2O4samples
从图2可知,锰源为人工研磨的样品Y0.5形貌较差,晶粒棱角不明显,颗粒尺寸差距较大,团聚形成的二次颗粒较多;与样品 Y0.5相比,锰源经2 h球磨处理的样品 Q2形貌发生了明显变化,晶粒尺寸更均匀,团聚形成的二次颗粒减少;样品Q4与Q8除了粒径分布更均匀外,晶粒的棱角也更清晰;样品Q16与Q32的晶粒更细小,原因可能是球磨较长。球磨处理的锰源合成的样品,粒径更加均匀,且随着球磨时间的延长,样品的晶粒尺寸逐渐变小。
2.2 XRD分析
合成的LiMn2O4样品的XRD图见图3。
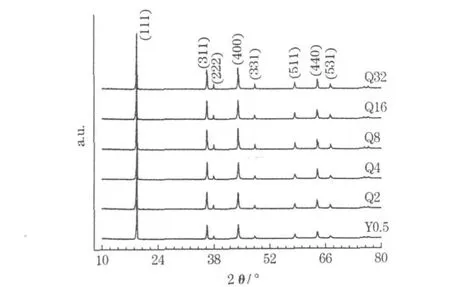
图3 合成的LiMn2O4样品的XRD图Fig.3 XRD patterns of prepared LiMn2O4samples
从图3可知,所有样品均为单一的尖晶石结构,未观察到其他相的衍射峰。随着球磨时间的延长,衍射峰变得更尖锐,说明结晶度逐步提高,原因是球磨使锰源的颗粒尺寸变小,更适于合成过程中的固相扩散。这与图1的结果一致。
合成的LiM n2O4样品的晶胞参数见表2。
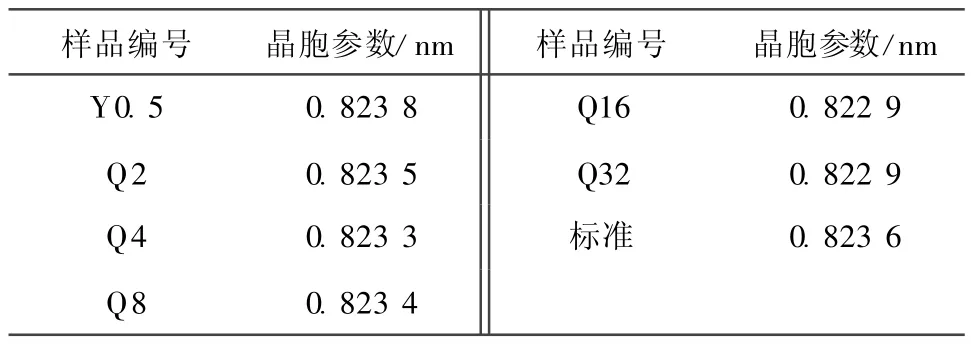
表2 合成的LiMn2O4样品的晶胞参数Table 2 Lattice parameters of prepared LiMn2O4samples
从表2可知,锰源为人工研磨的样品Y0.5晶胞参数较大;随着球磨时间的延长,晶胞参数整体呈下降趋势,表明随着球磨时间的延长,样品的晶体结构有所变化,锰源的粒径和形貌可能影响烧结时Li+/M n3+/Mn4+离子的占位[5]。
2.3 循环伏安测试
合成的LiM n2O4样品的循环伏安曲线见图4。
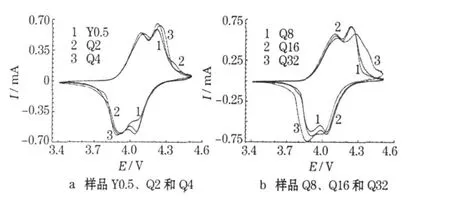
图4 合成的LiMn2O4样品的循环伏安曲线Fig.4 CV curves of prepared LiMn2O4samples
从图4可知,样品Y0.5氧化还原峰的范围较大,高电位的还原峰不明显,未体现出LiMn2O4材料应具有的特征双峰,表明Y0.5样品的结构有较大的缺陷;随着球磨时间的延长,样品Q2、Q4和Q8的氧化还原峰变窄,且双峰形状越发明显,表明合成的样品结构逐步完善,高于4.35 V的充电成分减少,电池的两个放电平台趋向理论值3.9 V和4.0 V。随着球磨时间进一步延长,与样品Q8相比,样品 Q16、Q32的充放电平台逐步变宽,且样品Q32的氧化还原峰有明显的畸变,表明过长的球磨时间会增加合成样品的结构缺陷。
2.4 恒流充放电测试
合成的LiM n2O4样品的首次放电曲线见图5。
从图5可知,锰源经球磨的样品首次放电比容量均大于110 mAh/g,且高于锰源为人工研磨的样品Y0.5。这主要是因为原料粒径的减小,缩短了热处理过程中的离子固相扩散距离,使产物的结构缺陷减少。
合成的LiMn2O4样品的循环性能见图6。
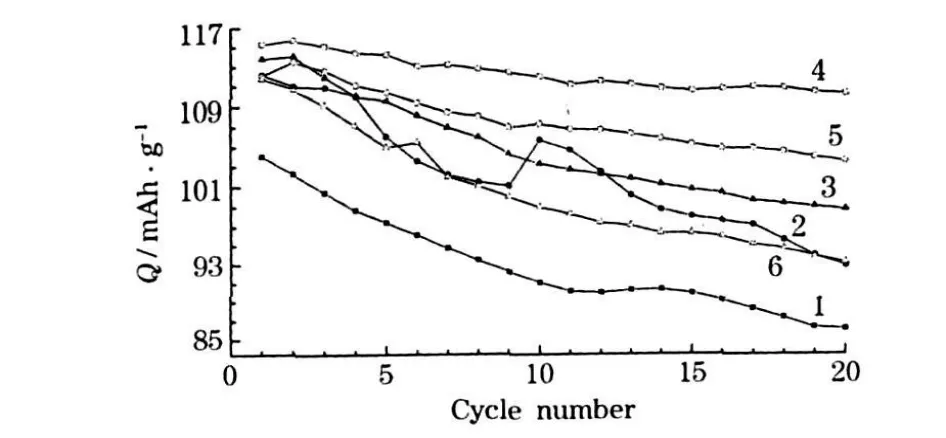
图6 合成的LiMn2O4样品的循环性能Fig.6 Cycle performance of prepared LiMn2O4samples
从图6可知,锰源经过8 h球磨的样品Q8具有最高的比容量和最好的容量保持率,首次及第20次循环的放电比容量分别为115.3 mAh/g和110.1 mAh/g,容量保持率为95.5%;锰源为人工研磨的样品Y0.5的比容量最低。样品Q2、Q4及Q8的比容量及循环性能整体上呈提高的趋势;而样品Q8、Q16及Q32的比容量和容量保持率逐渐降低。当EMD颗粒较大时,随着球磨时间的延长,晶粒尺寸逐渐减小和均匀,大颗粒及团聚形成的二次颗粒减少,因此在充放电过程中,晶粒表面与内部的 Mn价态差变小,晶粒表面的Jahn-Teller效应得到一定的抑制,尖晶石结构不易畸变,电化学循环稳定性增强;随着球磨时间进一步延长,产物的晶粒尺寸继续降低,使材料与电解液的接触面积增大,导致Mn在电解液中的溶解加剧,样品容量衰减加快。电化学循环的测试结果表明,EMD的球磨时间存在一个最佳值8 h。
3 结论
以颗粒较大的工业EM D为锰源,以LiOH为锂源,以熔融浸渍法合成了尖晶石相LiMn2O4。球磨处理EMD,对合成的LiMn2O4形貌和电化学性能的影响很大。在相同的烧结条件下,经球磨处理的EMD为锰源合成的LiMn2O4比以人工研磨的EMD为锰源合成的LiMn2O4的晶粒更均匀,且比容量及容量保持率更高。
球磨处理时间对合成的LiMn2O4的电化学性能有很大影响。以球磨8 h后的EMD作为锰源合成的LiMn2O4具有最好的电化学性能,锰源球磨处理时间过短或过长,均会导致合成的LiM n2O4正极材料的电化学性能降低。
[1]ZHANG Jin-jin(张瑾瑾),ZHOU You-yuan(周友元),ZHOU Yao(周耀),et al.LiMn2O4正极材料高温固相合成工艺的优化[J].Battery Bimonthly(电池),2010,40(1):27-29.
[2]Yoshio M,Inoue S,Hyakutake M,et al.New lithium-manganese composite oxide for the cathode of rechargeable lithium batteries[J].J Power Sources,1991,34(2):147-152.
[3]Xia Y,Takeshige H,Noguchi H,et al.Studies on an Li-Mn-O spinel system(obtained by melt-impregnation)as a cathode for 4 V lithium batteries(Ⅰ).Synthesis and electrochemical behaviour of LixMn2O4[J].J Power Sources,1995,56(1):61-67.
[4]Tu J,Zhao X B,Gao G S,et al.Enhanced cycling stability of LiMn2O4by surface modification with melting impregnation method[J].Electrochim Acta,2006,51(28):6 456-6 462.
[5]Nakayama M,Nogami M.A first-principles study on phase transition induced by charge ordering of Mn3+/Mn4+in spinel LiMn2O4[J].Solid State Commun,2010,150(29-30):1 329-1 333.
猜你喜欢
无机盐工业(2022年12期)2022-12-22 04:17:04
山东冶金(2022年4期)2022-09-14 08:59:00
选煤技术(2022年2期)2022-06-06 09:12:38
石材(2022年1期)2022-05-23 12:48:20
广东建材(2022年1期)2022-01-28 15:08:18
军事文摘(2020年18期)2020-10-27 01:55:10
石材(2020年2期)2020-03-16 13:12:44
中成药(2019年12期)2020-01-04 02:02:26
中国钼业(2018年5期)2018-11-02 07:40:18
凿岩机械气动工具(2016年3期)2016-03-01 04:00:24
