
Aβ诱导骨髓间充质干细胞对其分化的神经细胞Tau蛋白过度磷酸化的影响
2011-08-11 08:25赵天春韩雪飞
中风与神经疾病杂志 2011年7期
李 俊, 赵天春, 段 萍, 韩雪飞, 梁 晨, 邢 莹
β-淀粉样蛋白(β-amyloid,Aβ)的神经毒性是引起阿尔茨海默病(AD)神经元变性的重要因素,Aβ25-35是Aβ蛋白的主要的活性片段。骨髓间充质干细胞(MSCs)可以分化为神经细胞,应用于治疗神经系统疾病。在AD患者脑中的Aβ浓度高于正常,可以引起AD的另一个重要病理改变相关蛋白Tau磷酸化水平升高。Tau蛋白是微管的主要组成之一,Tau蛋白过度磷酸化会影响神经细胞胞体与轴突营养输送和细胞骨架的稳定。MSCs移植进入AD患者脑内,高浓度Aβ对其会有什么影响呢?我们的研究通过体外实验在高浓度Aβ环境中,诱导MSCs分化为神经细胞,观察体外诱导的神经细胞形态和骨架蛋白Tau的磷酸化的变化。
1 材料和方法
1.1 主要试剂 DMEM、胎牛血清(FBS)均购自Thermo公司。兔抗NSE抗体、GFAP抗体、PA免疫组化试剂盒为中杉金桥公司出品;鼠抗Tau抗体为USBiologieal公司产品;EGF和bFGF均为sigma公司产品。其余试剂均为国产分析纯级试剂。
1.2 动物 雄性SD大鼠,体重100g左右,普通级,由郑州大学实验动物中心提供。
1.3 MSCs的体外分离培养 SD大鼠断颈处死,放入75%酒精内浸泡5min。在无菌条件下,取出胫骨和股骨,灭菌的PBS缓冲液冲洗;剪断骨骺端,暴露骨髓腔,用加入10%FBS的L-DMEM培养液反复冲洗骨髓腔,得到的冲洗培养液吹打后,以1×106/ml接种到培养瓶,置于37℃、5%CO2的饱和湿度培养箱中培养。3d后首次换液,7d传代。以后每3~4d传代,第3代后细胞纯化后用于实验。
1.4 体外诱导MSCS向神经细胞分化及Aβ的干预 将细胞分为两组,对照组:不加Aβ,细胞达80%~90%融合时开始进行诱导分化,加入预诱导液DMEM培养基/体积分数为10%的FCS/10μg/L碱性成纤维生长因子(bFGF)培养24h,而后换为诱导液DMEM培养基/体积分数为2%(DMSO)二甲基亚砜/200μmol/L丁羟茴醚(BHA)诱导5h。诱导过程中在倒置显微镜下观察细胞形态变化。实验组:诱导分化前24h,在细胞培养基中加入Aβ(终浓度20μmol/L),其它同对照组。
1.5 Western blotting检测 弃去培养基,在培养瓶中加入胰酶消化,使细胞脱落,离心去上清,PBS洗去残留胰酶,将细胞移入0.5ml EP管,再加入100 μl细胞裂解液。震荡混匀,冰盒静置20min。放入低温离心机,4℃、12000r/min、10min,取出将上清移入另一新的干净0.5ml EP管,加入1/4体积5×蛋白加样缓冲液,混匀,沸水浴5min。取部分蛋白样品用BCA法进行蛋白浓度测定。根据测定的蛋白浓度,用1×蛋白加样缓冲液将各样本浓度调至一致。10%SDS-PAGE凝胶电泳(浓缩胶:恒压100V约20~30min,分离胶:恒压150V约 50~60min),湿转法转膜(恒压150V 60~80min),室温封闭1h,用封闭液稀释一抗,4℃孵育过夜,IgG二抗(1∶5000稀释),37℃孵育2h。ECL发光显色,X光底片曝光。以β-actin为内参照,实验重复3次。
2 结果
2.1 细胞形态观察及鉴定 骨髓间充质干细胞诱导分化神经样细胞前后细胞形态发生明显改变,有梭形分化为伸出突起的神经样形态(见图1、图2)。诱导后的实验组细胞突起明显短于对照组,且细胞折光性较差(见图3)。免疫细胞化学鉴定诱导后细胞表达NSE,即诱导后的细胞为神经细胞(见图4)。
2.2 Western blot Aβ实验组的诱导后神经细胞GSK-3β表达明显高于对照组细胞表达水平(见图5),Aβ 实验组 Tau[pSer262]蛋白和 Tau[pSer396]蛋白表达高于对照组诱导的神经细胞(见图 6、图 7)
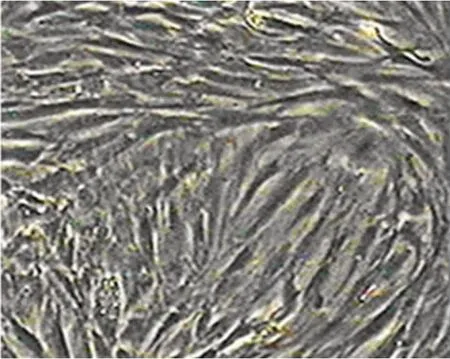
图1 诱导前骨髓间充质干细胞(40×)

图2 对照组诱导神经细胞(40×)

图3 Aβ处理组诱导神经细胞(40×)

图4 BMSC诱导后神经细胞的NSE染色(40×)

图5 GSK-3β表达
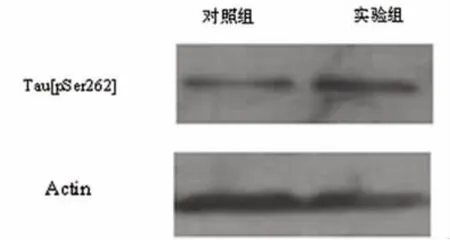
图6 Tau[pSer262]蛋白表达
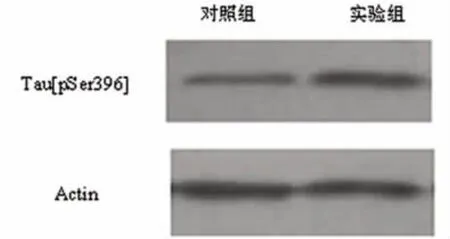
图7 Tau[pSer396]蛋白
3 讨论
Alzheimer病(AD)是近期记忆和智力功能进行性恶化为临床特点的一种神经系统常见疾病。AD大脑的特征性病理改变包括胞外Aβ沉积形成的老年斑和细胞内Tau蛋白异常磷酸化形成的神经元纤维缠结并伴随神经元的大量丧失[1]。因此,如果能补充AD大脑丢失的神经元,可以为AD的治疗找到一条新的途径。骨髓间充质干细胞具有分化为神经元细胞、胶质细胞的能力,能提供大量脑组织细胞的细胞。它可以作为细胞供体,对于治疗一些神经退行性疾病,具有重要的临床意义。
大量的实验结果和临床资料表明,Aβ是各种原因诱发AD的共同通路,是AD形成和发展的关键因素[2]。淀粉瀑布样反应假说认为AD患者脑中由于Aβ的沉积导致Tau蛋白被异常磷酸化,从而降低了微管组装能力,导致神经功能丧失。目前大多数学者认为Aβ是AD发病的始动因素,而Tau蛋白过度磷酸化可能是AD最重要的分子病理变化之一,是其发病机制中的一个核心环节[3]。
凝聚态Aβ25-35通过GSK-3β、MAPK等激酶可使正常的神经细胞 Tau蛋白异常磷酸化增多[4,5]。Tau蛋白有21个磷酸化位点,其中只有少数位点参与调节Tau蛋白的微管结合活性[6]。Tau蛋白在ser396、ser262位点的异常磷酸化具有一定的代表性。Hardy和Higgins认为Aβ可直接引起Tau蛋白过度磷酸化和NFTs的形成,最终导致神经元退行性改变[7]。Sigurdsson等将凝聚态 Aβ25-35注射至Fischer大鼠单侧杏仁核,观察到脑内杏仁核和海马神经元Tau蛋白异常磷酸化8d后增多,32d达到高峰,此后逐渐减弱,持续至128d。本实验结果显示Aβ可以促进骨髓间充质干细胞诱导的神经样细胞的Tau蛋白磷酸化水平升高,这与在神经组织的变化相同。
在Aβ的环境中骨髓干细胞可以分化为神经细胞,但细胞的突触明显比正常分化组细胞突触短。已有研究表明,在Tau缺陷的小鼠胚胎海马细胞培养与对照Tau蛋白正常表达的细胞对比,观察到缺陷小鼠细胞的轴突延伸延迟。说明Tau有促进轴突的生长的调节作用[8]。本实验发现Tau磷酸化水平升高,Tau蛋白促进轴突的延伸的作用减弱。另外Tau磷酸化后影响其与微管的结合,而不能起到稳定微管的作用[9],导致微管系统功能受损,进一步影响到轴浆运输,同样影响到轴突的延伸。
本研究表明,在Aβ的环境中,骨髓间充质干细胞分化为神经细胞受到明显影响,且严重影响细胞功能,故在AD患者采用骨髓干细胞移植时,应该采取相应措施以减少Tau异常磷酸化,例如移植细胞部位注入 GSK-3β抑制剂以减少 Tau蛋白磷酸化[10]。
[1]Castellani RJ,Rolston RK,Smith MA.Alzheimer disease[J].Dis Mon,2010,56(9):484-546.
[2]Ryan SD.Amyloid-beta42 signals Tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism[J].Proc Natl Acad Sci USA,2009,106(49):20936-20941.
[3]Hampel H.Total and phosphorylated Tau protein as biological markers of Alzheimer’s disease[J].Exp Gerontol,2010,45(1):30-40.
[4]Huang J.Unilateral amyloid-beta25-35 injection into the rat amygdala increases the expressions of aberrant Tau phosphorylation kinases[J].Chin Med J(Engl),2010,123(10):1311-1314.
[5]Sigurdsson EM.Local and distant histopathological effects of unilateral amyloid-beta 25-35 injections into the amygdala of young F344 rats[J].Neurobiol Aging,1996,17(6):893-901.
[6]Gendron TF,Petrucelli L.The role of Tau in neurodegeneration[J].Mol Neurodegener,2009,4:13.
[7]Hardy JA,Higgins GA.Alzheimer’s disease:the amyloid cascade hypothesis[J].Science,1992,256(5054):184-185.
[8]Morsch R,Simon W,Coleman PD.Neurons may live for decades with neurofibrillary tangles[J].J Neuropathol Exp Neurol,1999,58(2):188-197.
[9]Fath T,Eidenmuller J,Brandt R.Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease[J].J Neurosci,2002,22(22):9733-9741.
[10]Medina M,Avila J.Glycogen synthase kinase-3(GSK-3)inhibitors for the treatment of Alzheimer’s disease[J].Curr Pharm Des,2010,16(25):2790-2798.
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2021年10期)2021-12-02
家庭医药(2021年7期)2021-07-23
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年5期)2021-07-22
昆明医科大学学报(2021年2期)2021-03-29
现代临床医学(2021年2期)2021-03-29
世界科学技术-中医药现代化(2021年10期)2021-03-02
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
