
帕金森病大鼠胃肠功能障碍和肌间神经丛一氧化氮变化的研究
2011-08-11 08:25:50郭钢花臧卫东罗常月李倩倩
中风与神经疾病杂志 2011年7期
赵 静, 郭钢花, 臧卫东, 张 华, 罗常月, 李倩倩
帕金森病(Parkinson disease,PD)是以黑质多巴胺能神经元丢失为特征的运动障碍疾病,同时伴有胃肠功能障碍等非运动症状,主要包括吞咽困难、胃排空障碍和便秘[1]。其机制尚不明确,可能与胃肠肌间神经丛一氧化氮变化有关。Greene等[2]研究显示鱼藤酮诱导的PD大鼠胃肠传输功能受损,但肌间神经丛NO能神经元没有变化,Blandini等[3]研究显示6-OHDA诱导的PD大鼠伴随着结肠传输减慢,肌间神经丛NO能神经元是减少的,相反,Chaumette等[4]研究表明MPTP制作的灵长类PD模型肌间神经丛NO能神经元是增加的。为探讨PD肠神经系统一氧化氮的变化,本研究利用6-羟基多巴胺(6-OHDA)损毁大鼠单侧黑质多巴胺能神经元制作PD模型,观察PD大鼠肠神经系统NO的改变对胃肠功能的影响。
1 材料与方法
1.1 材料
1.1.1 实验动物 雌性 SD大鼠60只,体重180~220g,郑州大学实验动物研究中心提供。随机分为6-OHDA组和对照组,每组30只。
1.1.2 主要试剂与仪器 6-OHDA、阿朴吗啡(APO)(美国Sigma公司);兔抗大鼠一氧化氮合酶(NOS)Ⅰ型多克隆抗体(北京博奥森生物技术有限公司);免疫组化SP试剂盒、DAB显色试剂盒(北京中杉生物工程公司);逆转录(RT)试剂盒和聚合酶链(PCR)试剂盒(北京全式金生物技术有限公司);PCR引物由北京赛百盛生物技术有限公司合成。立体定位仪为江湾Ⅰ型;PCR GeneAmp System(2700)为美国Applied Biosystems公司产品;电泳仪(DYY-6C)为北京市六一仪器厂产品。
1.2 方法
1.2.1 大鼠 PD模型制作 10%水合氯醛(400mg/kg)腹腔注射麻醉,固定大鼠于立体定向仪上。以前囟为原点,参照包新民《大鼠脑立体定向图谱》确定右侧中脑黑质定位坐标(A:前囟后5.0mm,中线右侧旁2.4mm,硬脑膜下7.6mm;B:前囟后5.6mm,中线右侧旁开1.6mm,硬脑膜下 7.8mm)。钻开颅骨,向6-OHDA组大鼠的每个坐标点注射6-OHDA 3μl(4μg/μl,溶于含质量浓度为 0.02%的维生素 C的生理盐水中),以 1μl/min注药,留针10min,以1mm/min缓慢退针,随后消毒、缝合皮肤。对照组用上述方法向每个坐标点注射含0.02%维生素C的生理盐水3μl。术后3周,腹腔注射阿朴吗啡0.5mg/kg诱导大鼠向左侧旋转,记录30min内旋转次数,大于210r/30min的大鼠为成功模型。
1.2.2 1h粪便排出量 造模后28d将每只大鼠单独放在一个底部铺有滤纸的干净的笼子内。粪便排出后立即收集在一个密闭管中,称重后得到粪便湿重。在65℃下干燥12h,再次称重得到粪便干重。粪便含水量(%)=(粪便湿重-粪便干重)/粪便湿重×100%。
1.2.3 胃固体食物残留率测定 造模后28d,两组分别取10只大鼠禁食不禁水12h后,大鼠自由进食1h移去食物(进食前后分别称重食物)。2h后麻醉处死大鼠,取出胃用滤纸拭干后称全重,沿胃大弯剪开胃体,洗去胃内容物后用滤纸拭干,称胃净重,胃内残留率(%)=(胃全重-胃净重)/食物消耗量×100%。
1.2.4 胃肠nNOS免疫组化检测 造模后28d每组取10只大鼠麻醉后经4%的多聚甲醛灌流固定,剖腹取大鼠胃窦和近端结肠组织(距盲肠3cm以下),石蜡包埋,切片经脱蜡、梯度脱水后,3%H2O2室温孵育15min,微波修复抗原,10%山羊血清封闭10min,倾去血清,加一抗(兔抗鼠NOSⅠ型多抗),4℃孵育过夜,TBS洗3次,每次5min,加二抗37℃孵 30min,TBS洗 3次,每次 5min,DAB显色。苏木素复染,脱水、透明,中性胶封片,每组大鼠随机选10张相应位置的切片。采用Biosens Digital Imaging System v1.6分析软件计算胃窦和结肠nNOS免疫反应阳性区平均积分光密度。
1.2.5 胃窦及结肠平滑肌nNOS mRNA水平检测 造模后28d各组取10只大鼠麻醉处死后立即剖腹,常规Trizol法提取胃窦和近端结肠组织总RNA,用RT试剂盒进行反转录获取cDNA第一链片段。PCR扩增目的片段,nNO上游引物5’-CACATTTGCATGGGCTCG-3’,下游引物 5’-GACCTGAGATTCCCTTTGTT-3’;β-action 上 游 引 物 5’-CACCCGCGAGTACAACCTTC-3’下 游 引 物 5’-CCCATACCACCATCACACC-3’。反应条件为:94℃预变性 2min,94℃变性 30s,50℃退火 30s,72℃延伸2min,30个循环,全部周期结束后于72℃延伸6min。2%琼脂糖凝胶电泳,检测目的条带。用D-140图像记录分析系统对目的基因条带的平均灰度值与相应的内参条带的平均灰度值的比值进行比较。
2 结果
2.1 两组大鼠1h粪便排出量的比较
见表1。与对照组比较,6-OHDA组大鼠1h粪便排出量及粪便含水量显著低于对照组(P<0.01)。
2.2 两组大鼠胃固体食物残留率的比较
见表1。与对照组比较,6-OHDA组大鼠胃内固体食物残留率显著高于对照组(P<0.01)。
表1 两组大鼠1h粪便湿重、干重、含水量和胃固体食物残留率的比较(,n=10)

表1 两组大鼠1h粪便湿重、干重、含水量和胃固体食物残留率的比较(,n=10)
与对照组比较*P<0.01
组别 粪便湿重(g) 粪便干重(g) 含水量(%) 胃残留率(%)对照组6-OHDA组2.329 ±0.287 1.047 ±0.124*1.384 ±0.232 0.812 ±0.093*40.464 ±2.675 22.442 ±1.180*42.297 ±2.581 72.316 ±3.834*
2.3 两组大鼠胃肠肌间神经丛nNOS表达的比较
nNOS阳性产物(主要是nNOS阳性神经元及神经纤维)为棕黄色。6-OHDA组大鼠胃窦部和近端结肠肌间神经丛nNOS阳性区平均积分光密度分别是87.56 ±7.50 和104.11 ±5.22,显著低于对照组(132.02 ±11.91,142.82 ±6.32),差别有统计学意义(P <0.01)(见图1)。
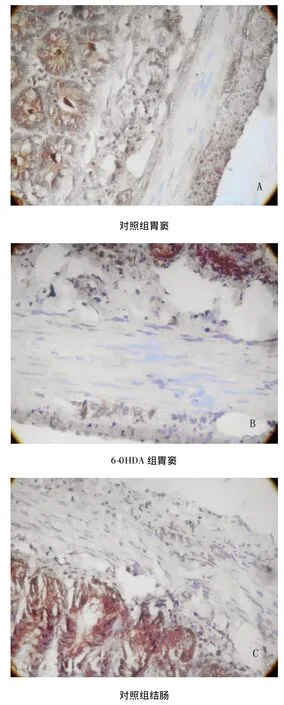
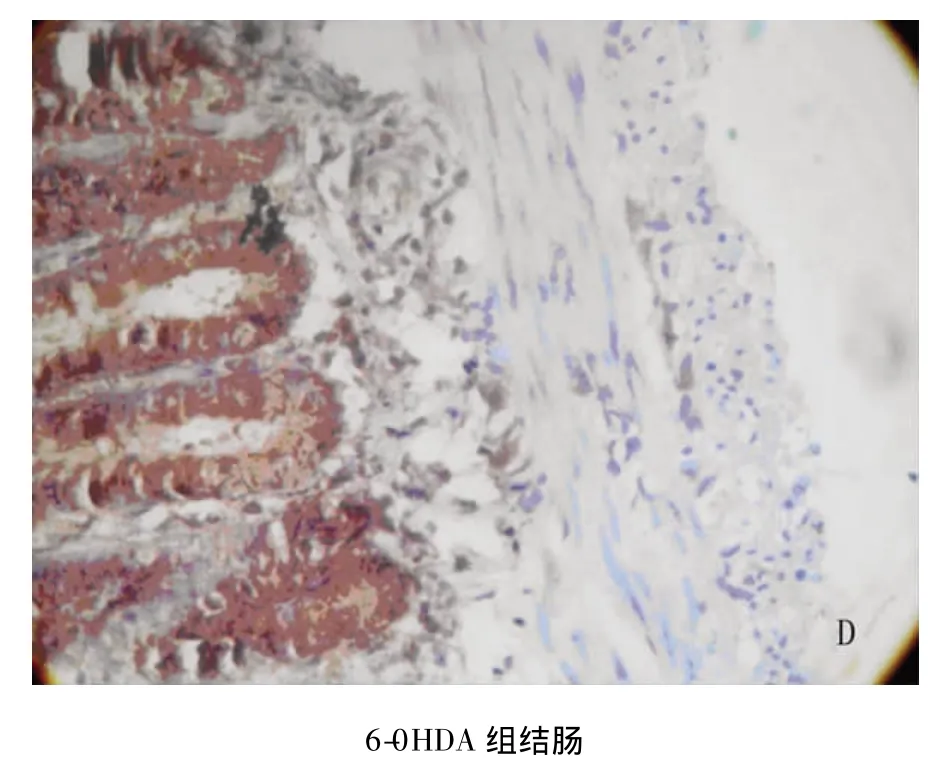
图1 6-0HDA组胃窦(B)及结肠(D)肌间神经丛nNOS表达明显低于对照组(A、C)(免疫组化染色,×400)
2.4 两组大鼠胃窦和结肠平滑肌nNOS mRNA水平的比较
应用RT-PCR方法得到的胃肠组织nNOS产物为386 bp,内参β-action为207 bp。6-OHDA组大鼠胃窦和近端结肠组织nNOS mRNA相对表达量为0.746±0.012 和 0.716 ±0.015,明显低于对照组(1.563 ±0.031 和 1.489 ±0.030,均 P <0.01)(见图2)。

图2 胃窦和结肠nNOS mRNA水平表达的变化
3 讨论
PD患者胃排空延迟主要表现为餐后胃胀气、早饱、恶心等,Edwards等[1]研究表明24%的PD患者有恶心症状,45%有胃胀气症状。Greene等[2]证明了鱼藤酮诱导的PD大鼠有明显的胃排空延迟;而1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)诱导的PD小鼠在不同的时间点胃固体食物的排空速度没有改变[5]。本研究表明6-OHDA组大鼠胃排空速度明显减慢,2h后胃内残留率明显大于对照组。
便秘是PD患者最常见的胃肠道症状。Jost等[6]发现所调查的PD患者中有80%结肠传输时间延长。Greene等[2]研究表明,鱼藤酮诱导的PD大鼠存在暂时性的排便频率减少;而MPTP处理的小鼠结肠运动功能亢进,排便频率增加[5]。本研究表明6-OHDA组大鼠在造模后第4周粪便排出量和粪便含水量较对照组明显减少。
PD是黑质纹状体多巴胺含量减少,乙酰胆碱系统功能相对亢进引起的运动障碍疾病,多巴胺和乙酰胆碱也是肠神经系统重要的神经递质。研究[2,4,5,7]表明 PD 患者和动物模型胃肠道胆碱能神经元数目没有改变,多巴胺能神经元数目明显减少。然而根据多巴胺抑制性的特点,胃肠道多巴胺能神经元的减少应该是加快而不是减慢胃排空。因此胃肠道非多巴胺能神经元在疾病进程中也可能受累。PD患者的肠肌丛,特别是NO能神经元发现存在asyn阳性包涵物,表明肠道NO能神经元受损可能与PD胃轻瘫和便秘有关。Greene等[2]研究表明鱼藤酮诱导的PD大鼠胃肠传输功能受损,但肌间神经丛NO能神经元没有变化。Anderson等[5]研究表明腹腔注射神经毒素 MPTP制作的PD小鼠模型,远端回肠NO能神经元没有变化,Blandini等[3]通过脑立体定向注射神经毒素6-OHDA诱导的大鼠PD模型结肠传输减慢伴随着肌间神经丛NO能神经元减少,也有研究[4]表明MPTP制作的灵长类PD模型肌间神经丛NO能神经元是增加的。本研究结果显示,6-OHDA组大鼠有胃排空延迟和便秘,胃窦和近端结肠肠肌丛nNOS免疫阳性神经元显著减少,nNOS mRNA水平显著降低。这些研究结果的差异可能是由于不同的PD动物模型NO能系统内在不同造成的,或者是由于不同的给药途径、给药剂量造成的。NO作为重要的抑制性神经递质主要作用是使胃肠道平滑肌发生松弛反应,缺乏可导致胃窦舒张受损和结肠持续性痉挛性收缩,引起胃排空延迟和结肠传输减慢。因此临床上PD患者出现的便秘和胃排空延迟可能是由于NO在胃肠道蠕动反射中介导的下行性抑制作用受损造成的。肠道NO能神经元受损可能会引起平滑肌过度收缩,导致肌肉痉挛引起结肠传输减慢,增加肠内容物水分的吸收[8],因此6-OHDA组大鼠粪便含水量较对照组显著减少。
本研究结果表明,6-OHDA诱导的PD大鼠存在胃排空延迟和便秘,可能与肠神经系统一氧化氮减少有关。目前,PD胃肠功能障碍还没有有效的治疗手段。研究PD肠神经系统神经递质的变化、探讨胃肠功能障碍的发生机制,可以早期进行更有效的药物干预治疗,提高患者的生活质量。
[1]Edwards LL,Pfeiffer RF,Quigley EMM,et al.Gastrointestinal symptoms in Parkinson’s disease[J].Mov Disord,1991,6:151-156.
[2]Greene JG,Noorian AR,Srinivasan S.Delayed gastric emptying and enteric nervous system dysfunction in the rotenone model of Parkinson’s disease[J].Exp Neurol,2009,218:154-161.
[3]Blandini F,Balestra B,Levandis G,et al.Functional and neurochemical changes of the gastrointestinal tract in a rodent model of Parkinson’s disease[J].Neurosci Lett,2009,467:203-207.
[4]Chaumette T,Lebouvier T,Aubert P,et al.Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism[J].Neurogastroenterol Motil,2009,21:215-222.
[5]Anderson G,Noorian AR,Taylor G,et al.Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson’s disease[J].Exp Neurol,2007,207:4-12.
[6]Jost WH,Schimrigk K.Constipation in Parkinson’s disease[J].Klin Wochenschr,1991,69:906-909.
[7]Singaram C,Ashraf W,Gaumnitz EA,et al.Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation[J].Lancet,1995,346:861-864.
[8]Ueki A,Otsuka M.Life style risks of Parkinson’s disease:association between decreased water intake and constipation[J].J Neurol,2004,251(7):18-23.
猜你喜欢
现代实用医学(2022年10期)2022-12-08 05:49:24
今日农业(2022年3期)2022-11-16 13:13:50
祝您健康·文摘版(2022年11期)2022-04-03 13:48:00
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:54:42
河南水产(2019年4期)2019-12-13 02:50:42
中国内镜杂志(2017年2期)2017-03-20 16:18:10
妈妈宝宝(2017年2期)2017-02-21 01:21:28
海洋世界(2017年1期)2017-02-13 08:31:53
医学研究杂志(2015年11期)2015-06-10 06:44:03
吉林医学(2014年18期)2014-08-15 00:53:03
