
固元胶囊多糖的分离纯化与初步结构研究
2011-07-28 05:25陈卫平陈琴鸣戴德雄刘根才
中国药业 2011年23期
陈卫平,陈琴鸣,刘 斌,戴德雄,刘根才
(1.浙江省丽水市食品药品检验所,浙江 丽水 323000; 2.浙江维康药业有限公司,浙江 丽水 323000)
固元胶囊是一种免疫调节剂,用于癌症患者经化学治疗或放射治疗引起的免疫功能和造血功能障碍,主要由黄芪粗多糖和人参芦头组成(黄芪粗多糖∶人参芦头为 216.7 g∶55.7 g),黄芪粗多糖为主药,且为发挥免疫调节作用的主要成分。固元胶囊质量标准中仅以硫酸-苯酚法测定多糖总量来控制原料和成品的质量,无相对分子质量与分布、单糖组成测定项目,不能有效地控制固元胶囊的质量。笔者对固元胶囊中的多糖组分进行分离纯化,得到精制品,并进行了初级结构的初步研究,为进一步提高固元胶囊的质量标准提供依据。
1 仪器和材料
Agilent 1100型高效液相色谱仪(美国安捷伦公司);Alltech-2000ES型蒸发光散射检测器(美国奥泰科技公司);TU-1901型紫外-可见分光光度计(中国普析通用公司);FTIR-8400S型红外分光度计(日本岛津公司)。鼠李糖(Rha)、阿拉伯糖(Ara)、木糖(Xyl)、甘露糖(Man)、半乳糖(Gal)、葡萄糖(Glc)、葡萄糖醛酸(Glc A),均购自上海楷洋生物技术有限公司,含量不低于99.0%;氮气(99%);固元胶囊(浙江维康药业有限公司,批号为20070202);乙腈、甲醇为色谱纯,纯净水,其余试剂为分析纯。
2 方法和结果
2.1 固元多糖的提取、精制[1]
将固元胶囊的内容物研细混匀,称取10 g,加水200 mL微热使溶解,3500 r/min离心10 min,取上清液,加水200 mL,重复操作,合并上清液,加30% 的H2O2溶液60 mL于50℃水浴保温3 h,至溶液变为淡黄色半透明溶液后,置水浴中边搅拌边缓慢加入等体积10%的三氯醋酸,于冰箱内过夜,3500 r/min离心6 min,取上清液,置旋转蒸发仪上浓缩至约50 mL,缓缓加入5倍体积的无水乙醇,放置冰箱过夜,经4号砂芯漏斗过滤,沉淀依次用无水乙醇、丙酮洗涤3次,真空干燥5 h,得白色的固元多糖精制品粉末。
2.2 纯度检查
紫外分光光度法:取多糖精制品适量,加蒸馏水制成1 g/L的溶液,于200~400 nm波长范围内进行紫外扫描。结果未发现有核酸(260 nm)及蛋白质(280 nm)的特征吸收峰。
高效分子排阻色谱(HPSEC-ELSD)法:取多糖精制品适量,用水制成1 g/L的溶液,高速离心后用0.45 μm微孔滤膜过滤,取30 μL注入色谱仪,用高效分子排阻色谱法分析。结果可见3个色谱峰,表明其由3部分组成,色谱图见图1。
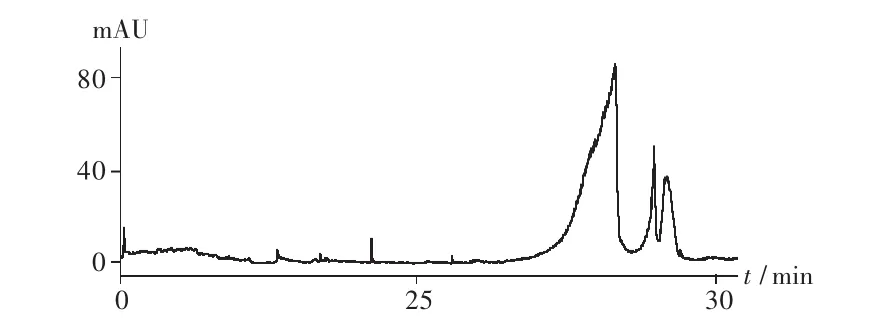
图1 固元胶囊多糖精制品高效分子排阻色谱图
2.3 固元多糖各组分的分离
取上述固元多糖精制品,以水为洗脱剂,用Sephadex G 150填充柱洗脱,用高效分子排阻色谱法检测,收集相同组分。60℃减压浓缩至小体积,加6倍体积无水乙醇沉淀,沉淀按2.1项下方法洗涤、干燥,得到纯品A,而B,C两组分比例极少,未得到沉淀。固元胶囊多糖A组分的高效分子排阻色谱图见图2。
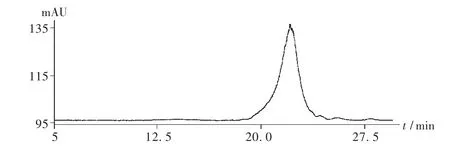
图2 固元胶囊多糖组分高效分子排阻色谱图
2.4 固元胶囊多糖A组分的结构分析
理化性状[2]:固元胶囊多糖A组分为白色粉末,溶于水(尤其是热水),而不溶于乙醇、乙醚、丙酮和氯仿,水溶液微有乳光,苯酚-硫酸反应呈阳性,比旋度[a]D20=+130°。
相对分子质量及其分布测定:取固元胶囊多糖A组分,用水制成1 g/L的溶液,高速离心后用0.45 μm微孔滤膜过滤,取20 μL注入色谱仪。结果见表1。
单糖组成测定:取固元胶囊多糖A组分,经水解,用色谱柱Prevail Carbohydrate ES柱,流动相CH3CN-H2O(80∶20),流速 0.8 mL/min,载气体(N2)流速为2.2 L/min,漂移管温度为82.5℃,测定单糖。结果A 组分由 Glc A,Rha,Ara,Xyl,Gal,Glc 组成,摩尔比为22.3 ∶0.04 ∶0.08 ∶0.09 ∶1。色谱图见图 3。

表1 固元胶囊多糖A组分的相对分子质量测定结果(n=3)

图3 样品和对照品混合溶液高效液相色谱图
红外光谱分析:取纯多糖固元胶囊多糖A组分约20 mg,以KBr压片,由FTIR-8400 S型红外分光度计扫描红外光谱,见图4。结果在3600~3200 cm-1有宽展圆滑强吸收峰,为糖类-OH伸缩振动;3000~2800(2933)cm-1处吸收峰为糖类 CH3、CH2和CH等的C-H伸缩振动;在1665~1625(1636)cm-1处有一组吸收峰,为糖的水化物的吸收峰;1485-1445 cm-1的吸收峰为CH的弯曲振动;1250~950 cm-1之间的一组峰是两种C—O的伸缩振动吸收峰;1030 cm-1的吸收峰为吡喃环结构的C—O振动,892 cm-1有极弱的吸收峰说明含少量的β糖苷键;833 cm-1有吸收峰,说明是α糖苷键;762 cm-1为D-葡萄吡喃糖环对称环伸缩振动;1650~1550 cm-1处无N-H变角振动的吸收峰,说明无一级和二级氨基;810 cm-1无吸收峰说明无甘露糖。

图4 固元胶囊多糖A组分的红外光谱图
高碘酸氧化及Smith降解[3]:称取固无胶囊多糖A组分25 mg,用少量水溶解,多糖组分固元胶囊多糖A组分经高碘酸氧化反应,144 h后高碘酸消耗量达最大,平均每摩尔已糖残基消耗0.61 mol高碘酸,同时无甲酸生成。根据高碘酸氧化原则,以1→2或1→4位键合的糖基经高碘酸氧化,平均每个糖基仅消耗1分子高碘酸,且无甲酸释放;以1→3位键合的糖基不被高碘酸氧化;以1→6键合的糖基或非还原末端基经高碘酸氧化,消耗2分子高碘酸,同时释放1分子甲酸;以1→3位键合的糖基不被高碘酸氧化。因而可推知,固元胶囊多糖A组分不含或极少含1→6位键糖苷,必含1→2或1→4位键糖苷。经Smith降解后的薄层色谱检出甘油和乙二醇,但无赤藓醇,乙二醇可能为制备过程中带入,可推知固元胶囊多糖A组分为1→2位糖苷键,可能还有少量1→4糖苷键。
3 讨论
对组分固元胶囊多糖A组分的结构分析表明,其为由Glc A等5个单糖组成的酸性杂多糖,由薄层色谱法与非衍生化高效液相色谱-蒸发光散射检测法检测结果基本相同,其相对分子质量用高效分子排阻色谱法测定,结合红外光谱、高碘酸氧化和Smith降解结果,可推测该组分结构中单糖的连接点类型。结果表明,固元胶囊多糖A组分的比旋度为[a]D20=+130°,重均分子量为1.62 × 106,由 Glc A,Rha,Ara,Xyl,Gal,Glc 组成,物质的量之比为22.3 ∶0.04 ∶0.08 ∶0.09 ∶1,以 α-(1→2)型葡萄糖苷键构成糖链,可能还有少量β-(1→2)糖苷键。
[1]YBZ04242005,国家食品药品监督管理局标准(试行)[S].
[2]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:附录 40.
[3]张维杰.糖复合物系列化研究技术[M].第2版.杭州:浙江大学出版社,1999:168.
猜你喜欢
食品与生物技术学报(2022年1期)2023-01-11
煤气与热力(2021年12期)2022-01-19
中国免疫学杂志(2019年17期)2019-09-20
中成药(2018年8期)2018-08-29
天然产物研究与开发(2018年6期)2018-07-09
中成药(2018年2期)2018-05-09
中成药(2018年3期)2018-05-07
分析化学(2017年5期)2017-06-21
中成药(2014年11期)2014-02-28
中成药(2014年11期)2014-02-28
