
非晶红色荧光粉Cd3Al2Si3O12∶Eu3+的制备及发光性能
2011-07-04 01:18:50刘东平牛金海
无机化学学报 2011年8期
丛 妍 李 斌 刘东平 杨 杞 牛金海
(1大连民族学院理学院,大连 116600)(2中科院长春光学精密机械与物理研究所激发态重点实验室,长春 130033)
InGaN蓝光二极管(LED)激发的白光LED在固体照明领域中有诸多优点,如高效率,低能耗,高可靠性以及寿命长等,使其成为人们研究的焦点。白光LED将替代传统的白炽灯和荧光灯,应用于移动电子产品的背景照明,医疗和生活照明等领域[1-2]。白光LED的商业化已经通过470 nm的蓝光LED与宽带发射的YAG∶Ce3+的黄光磷光体的复合得到了实现[3]。但是,这种复合得到的白光LED由于红光成分(>600 nm)的缺乏,使得得到的白光的显色指数较差(<80)[4]。另外一个途径得到白光就是通过近紫外光LED(400 nm左右的发射)作为激发光源与红、绿、蓝三基色相复合[5-6]。无论哪种方法,要想得到色饱和度较好的白光,都需要有波长较长的红光成分。到目前为止,常见的白光LED用红色磷光体的研究主要集中在氮化物上。氮化物具有高的发光效率和好的热稳定性,使其成为具有应用潜力的白光LED用红色荧光粉[7]。但是,由于缺乏合适的合成方法,高的合成成本的限制以及原料的化学敏感性等原因,在过去的几年时间的研究中只得到了少量的氮化物[8-9]。因此,寻找易合成的,高效的红色荧光粉成为白光LED发展的迫切需要。
稀土元素因其特有的4f电子结构,发射光为线谱或窄带谱的优良性能,在荧光粉中倍受青睐。稀土离子常常在发光材料中作为发光中心,例如,彩色显像管中的红粉普遍使用的是铕激活的硫氧化钇(Y2O2S∶Eu3+),YVO4∶Eu3+却常常作为高压汞灯中的红色荧光粉。但是这些传统的荧光粉在近紫外区激发的光效不高,并且传统的硫化物荧光粉的稳定性差,制备过程容易污染环境。
本工作使用溶胶-凝胶法合成了一种新的Eu3+掺杂的Cd3Al2Si3O12非晶体系红色荧光材料。该红色荧光粉可以被近紫外(394 nm)和蓝光(464 nm)有效激发,制备简单,热稳定性好。
1 实验部分
1.1 样品的合成
称取分析纯硝酸镉(Cd(NO3)2·4H2O)和硝酸铝(Al(NO3)3·9H2O),溶于乙醇中,然后加入正硅酸乙酯(TEOS)作为SiO2的来源,在搅拌下加入水和少量硝酸进行水解,调节pH值为2左右。最后的nTEOS∶nEtOH∶nH2O=1∶12∶4。再以硝酸铕(Eu(NO3)3)的形式加入 Eu3+离子,掺杂物质的量分数范围为10%~40%。将上述混合溶液搅拌2 h得到透明溶胶,溶胶在60℃下烘干1~2 d得到凝胶。将得到的干凝胶研磨成粉末状装入坩埚内,在500~900℃下灼烧3 h,空气中冷却,得到白色粉末。
1.2 样品的表征
样品的X射线衍射(XRD)图在日本Rigaku D/max-IIB衍射仪上测量,测试电压为40 kV,电流为20 mA,扫描步宽为 0.02°(2θ),采用 Cu 靶 Kα1 辐射线(λ=0.154 06 nm)作为辐射源。以氙灯作为激发源,用F-4500荧光光谱仪测量样品的室温激发和发射光谱。
2 结果与讨论
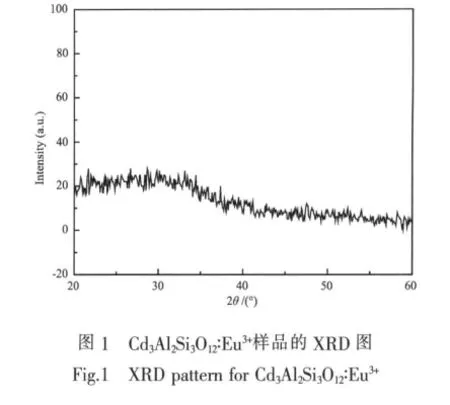
Cd3Al2Si3O12∶Eu3+荧光粉是一个非晶体系,从图1给出的烧结温度在800℃时获得样品的XRD图就可以证明。从图中可以看出,Cd3Al2Si3O12的衍射图中并没有出现任何晶体衍射峰,表明在样品中并没有形成任何晶相,而是以非晶形式存在的。当烧结温度达到900℃时,样品呈现玻璃态。这种无定形非晶结构有利于大浓度的Eu3+离子掺杂。
图2展示了在800℃下烧结的Cd3Al2Si3O12∶Eu3+样品监测611 nm位置发射的室温下的激发光谱。其激发光谱展示了 Eu3+的7F0基态到5HJ,5D4,5GJ,5L6,5D3,2,1激发态的跃迁。由200延伸到300的宽带对应的应该是Eu3+的电荷迁移带(CTB)。如图所示,最强的吸收峰是位于394 nm的7F0→5L6和位于464 nm的7F0→5D2跃迁,以及一个中心位于254 nm的电荷迁移带。394和464 nm的位置与紫外和蓝光LED的发射位置相符合。因此,Cd3Al2Si3O12∶Eu3+非晶荧光粉可被近紫外和蓝光LED有效激发。
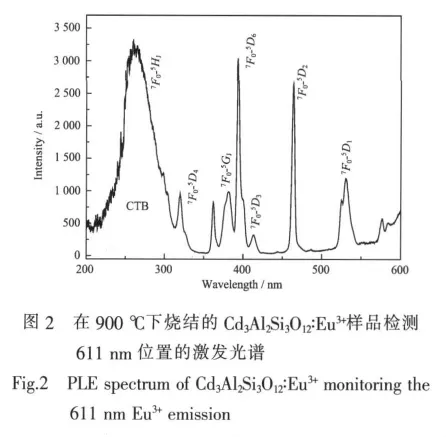
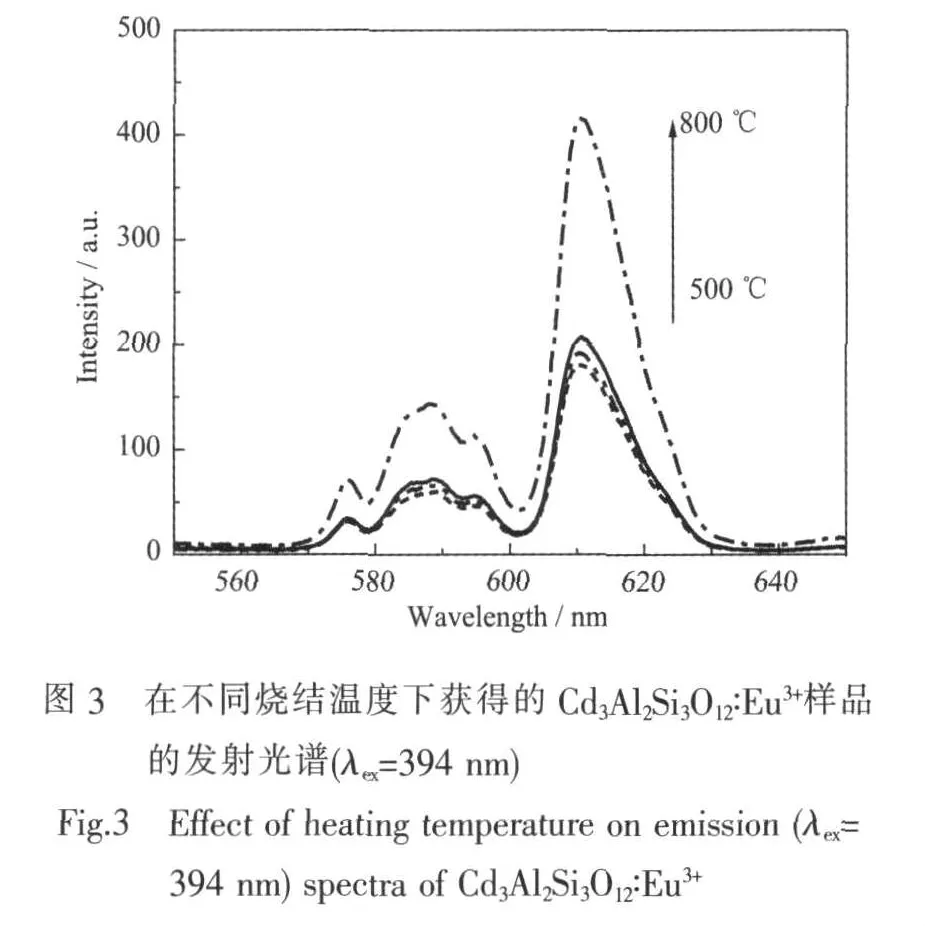
图3是从500到800℃的烧结温度下获得的不同 Cd3Al2Si3O12∶Eu3+样品室温下的发射光谱。在394 nm紫外光的激发下,样品的发射光谱包含了一个位于611 nm的Eu3+的源于5D0→7F2跃迁的红光发射以及一个较弱的源于5D0→7F1跃迁的590 nm的发射。Eu3+的5D0→7F1是磁偶级跃迁,几乎不受外界的晶场环境的影响。5D0→7F2是电偶级跃迁,对格位对称性非常敏感。要想得到色纯度较好的红光发光材料,就要求发射集中在5D0→7F2跃迁,即610 nm左右[10-11]。如图 3所示, 样品的5D0→7F2跃迁的611 nm发光要比位于590 nm的7F1能级的发光强的多。这与XRD的结果相符合,证明了Eu3+的加入并没有改变材料非晶结构,非晶体系的对称性较低,Eu3+偏离对称中心。如果Eu3+的掺杂浓度足够高的话,就会产生如下的交叉弛豫[12]:

如图3所示,样品的发光强度随着烧结温度的升高而升高。亮度的提高一部分归结于在高温下的基质材料的脱水。在湿化学反应方法制备的样品中极容易引进-OH离子基团,-OH键的震动主要发生在2 700~3 700 cm-1的范围[13],所以只有很少数的声子用来作为5D0能级的无辐射激发。因此,-OH离子被认为是通过多光子弛豫对Eu3+发光产生严重猝灭的原因。样品的发光强度从500到700℃的变化很不明显,当烧结温度升高到800℃时,发光强度明显的增加。这表明-OH基团是在烧结温度达到800℃时被比较彻底的除掉的。
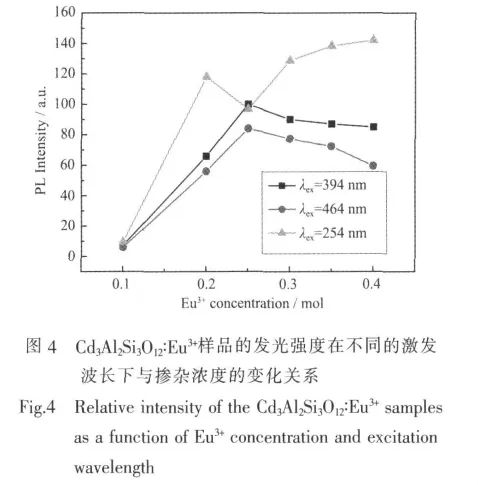
Cd3Al2Si3O12∶Eu3+非晶荧光体的 Eu3+的特征红光最大优点就是在394和464的位置有强的吸收。在Eu3+的掺杂浓度为0.25 mol时,样品在394 nm的激发下的发射强度几乎与在254 nm位置的电荷迁移带激发下是一样的,在464 nm的激发下的发光强度略有降低,大约是在254 nm激发时的80%。图4展示了样品的发光强度与样品的掺杂浓度和激发波长的关系。从图中我们可以看出,Cd3Al2Si3O12∶Eu3+样品在不同的激发波长的激发下有着不同的猝灭浓度。在254 nm的激发下,样品的发光强度随着掺杂浓度的增加而增强,在试验范围内没有观察到固定的猝灭浓度。因为材料非晶体系的特殊结构,在基质中可以掺杂较高浓度的激活剂离子。而在394和464 nm的激发下,当掺杂浓度达到0.25 mol时,样品的发光强度分别都达到了最大。当掺杂浓度高于0.25 mol时,由于浓度的猝灭,发光强度开始降低。因此,非晶荧光粉Cd3Al2Si3O12∶Eu3+可以被近紫外(394 nm)和蓝光(464 nm)有效的激发获得红光,使得这种荧光粉可以在紫外和蓝光发光二极管的激发下与蓝、绿色荧光粉复合获得白光LED。
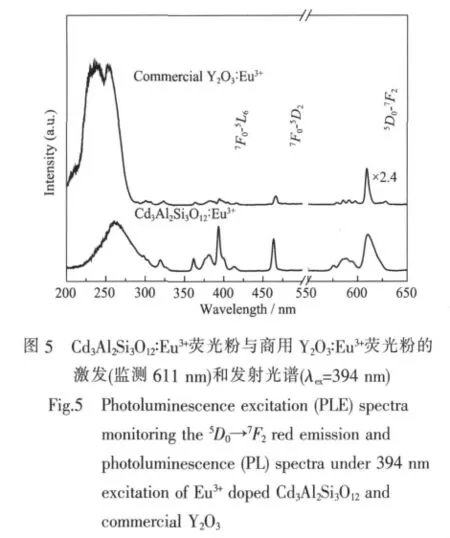
我们还把Cd3Al2Si3O12∶Eu3+红色荧光粉与传统的商用Y2O3∶Eu3+红色荧光粉进行了比较。如图5所示,Y2O3∶Eu3+在7F0→5L6(394 nm)和7F0→5D2(464 nm)2 个位置有2个弱的激发峰和1个强的电荷迁移带。而在 Cd3Al2Si3O12∶Eu3+荧光粉中,在7F0→5L6(394 nm)和7F0→5D2(464 nm)2个位置都有很强的吸收峰(大约是Y2O3∶Eu3+的10倍)。图的右边是在394 nm激发下的发射光谱,Cd3Al2Si3O12∶Eu3+的位于 611 nm 位置的Eu3+的特征发光是 Y2O3∶Eu3+的 2.4 倍左右。
考虑到LED在工作时温度将上升,能够用于白光LED的荧光粉必须要具有好的热稳定性,在连续使用产生高温以后,对发光性能不能有太大的影响[14]。因此,测定了 Cd3Al2Si3O12∶Eu3+从室温 30 ℃升温到130℃的温度猝灭,并与已经商业化的白光LED中常用的YAG∶Ce3+荧光粉(自制)进行了比较。我们从图6中可以看到,当温度从室温(30℃)升高到130℃,YAG∶Ce3+样品的发光强度降到初始值的76%。然而,在我们的 Cd3Al2Si3O12∶Eu3+样品中,发光亮度并没有任何下降,并且略微升高到了初始值的116%。发光二极管的结温一般低于127℃,因此,即使结温的温升达到127℃,或者二极管内的反射杯的温度达到127℃时,对Cd3Al2Si3O12∶Eu3+的发光强度影响是较小的。因此Cd3Al2Si3O12∶Eu3+样品的结构比YAG∶Ce3+样品更加稳定,对热猝灭的影响更小。

3 结 论
使用溶胶-凝胶法制备了 Cd3Al2Si3O12∶Eu3+非晶体系红色荧光粉,并对其发光性质进行了研究。结果表明,该荧光粉在位于394 nm的5L6能级和464 nm的5D2能级的激发下猝灭浓度为0.25 mol,产生强的Eu3+的位于611 nm的5D0→7F2电偶极跃迁为主的特征红光发射。随着烧结温度的升高,对Eu3+的发光有起猝灭作用的OH-离子基团被移除,使得Eu3+的611 nm发光逐渐增强。与传统的荧光粉相比较,Cd3Al2Si3O12∶Eu3+荧光粉的发光强度强(与 Y2O3∶Eu3+相比较),热稳定性好(与 YAG∶Ce3+相比较)。
[1]Nakamura S,Fasol G.The Blue Laser Diode.Berlin:Springer,1997:343
[2]Yum J,Seo S,Lee S,et al.J.Electrochem.Soc.,2003,150:H47-52
[3]Lee S,Seo S.J.Electrochem.Soc.,2002,149:J85-88
[4]Ponce F,Bour D.Nature,1997,386:351-359
[5]Huh Y,Shim J,Kim Y,et al.J.Electrochem.Soc.,2003,150:H57-60
[6]Narukawa Y,Niki I,Izuno K,et al.J.Appl.Phys.,2002,42:L371-373
[7]Mueller-Mach R,Mueller G,Krames,et al.Phys.Status Solidi A,2005,202:1727-1732.
[8]Schlieper T,Milius W,Schnick W.Anorg.Allg.Chem.,1995,621,1380-1384
[9]Piao X,Horikawa T,Hanzawa H,et al.Appl.Phys.Lett.,2006,88(16),161908-1-3
[10]Judd B.Phys.Rev.,1962,127:750-761
[11]Ofelt G.J.Chem.Phys.,1962,37:511-520
[12]Kitai A.Solid State Luminescence,London:Chapman Hall,1993:38
[13]Zhang L,Hu F.J.Phys.Chem.Solid.,2002,63:575-579
[14]Muthu S,Schuurmans F,Pashley M.IEEE J.Quantum Electron.2002,8:333-335
猜你喜欢
陶瓷学报(2021年5期)2021-11-22 06:34:58
电镀与环保(2017年2期)2017-05-17 03:42:18
照明工程学报(2016年3期)2016-06-01 12:18:01
连环画报(2016年4期)2016-05-05 13:51:25
中国房地产业(2016年9期)2016-03-01 01:26:18
电源技术(2016年2期)2016-02-27 09:04:39
大连工业大学学报(2015年4期)2015-12-11 04:06:50
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:30
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:30
中国科技信息(2015年21期)2015-11-07 08:41:50
