
胶原Ⅲ肾病
——形态学特点和临床表现
2011-07-01 21:59陈惠萍黄高渊谢红浪朱茂艳张明超梁少姗曾彩虹刘志红
肾脏病与透析肾移植杂志 2011年6期
陈惠萍 徐 峰 黄 倩 黄高渊 谢红浪 朱茂艳 张明超 贺 倩 梁少姗 曾彩虹 刘志红
胶原Ⅲ肾病
——形态学特点和临床表现
陈惠萍 徐 峰 黄 倩 黄高渊 谢红浪 朱茂艳 张明超 贺 倩 梁少姗 曾彩虹 刘志红
目的:总结一种非免疫因素介导的肾小球疾病——胶原Ⅲ肾病的肾活检形态学及实验室检查特点,旨在提高对此病的诊断水平。 方法:回顾性分析经肾活检免疫荧光、电镜检查确诊胶原Ⅲ肾病的12例患者的组织学特点,并分析其临床表现及实验室检查结果。 结果:12例患者男性4例,女性8例,病程3~300月(平均62.08±60.96月);年龄21~67岁。蛋白尿为本组患者最常见的肾脏损害首发症状(10例,83.33%),10例(83.33%)患者病初即诊断高血压,尿沉渣红细胞计数>50万/m l者仅1例(8.33%)。肾小管受损的实验室指标包括尿NAG酶升高9例(75%);禁水13h尿渗量减低者9例。确诊时血清肌酐(SCr)升高者4例,7例贫血。肾活检免疫荧光检查C3阳性者5例(41.67%),3例(25%)患者合并IgA阳性,胶原Ⅲ染色证实非废弃或非硬化部位的肾小球外周袢和(或)系膜区弥漫阳性荧光;组织学观察证实,12例患者肾小球体积均增大,且肾小球内皮下疏松、区域增宽等,肾小球病变大致分为两种类型,一类肾小球非增生性的“结节样”改变,PAS或PASM-Masson染色肾小球毛细血管袢内皮下呈均质、淡染的嗜复红或嗜亮绿色;另种“结节样”改变不明显,肾小球细胞总数不多或增加,内皮细胞成对,PASM-Masson染色外周袢弥漫双轨。超微结构观察证实12例患者肾小球毛细血管袢内皮下疏松、区域增宽,内皮下和(或)系膜区见束状或单枝的、有明暗带、直径约43~60 nm的胶原纤维,3例(25%)免疫荧光染色IgA阳性者在肾小球系膜区见团块状的电子致密物沉积。随访过程中病初4例(33.3%)SCr升高者未降至正常,病初SCr正常者中仅1例在随访中轻度增高。 结论:高血压和蛋白尿是胶原Ⅲ肾病最常见的临床表现,肾活检组织学改变具特征性,诊断需借助免疫荧光染色和超微结构观察;预后相对良好。
胶原Ⅲ肾病 肾活检 高血压 蛋白尿
1979年日本学者首次报道一种以肾病综合征或大量蛋白尿为首发临床症状的新型肾小球疾病,其临床经过迁延、病情进展缓慢,最终可进展至肾功能衰竭[1]。由于电镜观察证实肾小球基膜(GBM)内皮下区域和系膜区存在大量胶原纤维,1990年,经免疫荧光染色证实电镜观察胶原纤维为Ⅲ型胶原[2],1991年,有学者建议将其命名为 “Ⅲ型胶原肾小球病”,简称“胶原Ⅲ肾病”[1];1995年世界卫生组织将其收录进肾小球疾病的分类中[3]。
该病较少见,欧洲、巴西仅有零散病例报告[4],多数患者来自日本,我国目前报告的患者数不足20例。回顾分析发现,90年代初我们曾见到病理特征高度类似胶原Ⅲ肾病的患者,由于认识不足,临床资料及实验室检查结果也不完整,更未进行仔细的超微结构观察和免疫荧光Ⅲ型胶原染色,因而未能明确诊断。近年来随着医疗水平进步,我们发现该病虽相对少见但并非罕见。本文报道12例临床、实验室检查和病理资料完整的胶原Ⅲ肾病患者,以提高对本病的认识。
对象和方法
病例选择 在南京军区南京总医院全军肾脏病研究所住院行肾活检者,经肾活检免疫病理、超微病理和组织学检查符合胶原Ⅲ肾小球病,排除膜增生性肾小球肾炎、冷球蛋白血症肾损害、病毒感染相关的肾脏疾病及狼疮性肾炎等疾病。12例患者中男性4例,女性8例;病程约3~300月[平均(62.08± 60.96)月,中位时间18月];肾活检时年龄21~67岁(平均43±13岁)。
临床观察指标 肾脏损害的病程、首发临床症状、肾脏病家族史、肝肾功能、血电解质、冷球蛋白、自身抗体、24h尿蛋白定量、尿沉渣红细胞计数;肾小管受损的实验室指标(包括尿NAG,尿RBP及禁水13h尿渗量)。
肾脏B超检查 测量两侧肾脏长径、宽度和厚度。
临床相关指标定义 高血压定义:收缩压≥130 mmHg和(或)舒张压≥90 mmHg;肾病综合症定义:尿蛋白定量≥3.5 g/d,血清白蛋白<30 g/L;肾功能不全定义:血清肌酐(SCr)>109.62μmol/L;终末期肾病(ESRD)定义:SCr>707.2μmol/L;贫血定义:血红蛋白(Hb)<110 g/L。
肾脏病理 全部患者均在B超引导下行经皮肾穿刺活检术。肾穿刺取得的组织分成三部分,按常规方法分别进行光镜、电镜及免疫荧光检查。
光镜 标本以甲醛液或酒精-苦味酸-甲醛液固定,石蜡包埋后连续切片,组织切片厚1.5~2μm,分别行HE、PAS、PASM-Masson,Masson-Trichrome染色。
免疫荧光 采用冰冻切片,切片厚3μm,以异硫氰酸荧光标记的羊抗人荧光抗体行直接法染色,检测肾组织中IgG、IgA、IgM和补体C3、C4、C1q沉积的部位、强度和分布特点。
胶原Ⅲ染色:石蜡切片脱蜡至水,0.1%胰酶消化5 min,加一抗fibronectin(兔多克隆抗体. DAKO),1∶400室温过夜;PBS冲洗,加FITC二抗(猪抗兔.DAKO),室温孵育40 min,清水冲洗,吹干,甘油封片置荧光显微镜下观察。
电镜 肾组织以3.75%的冷戊二醛固定,1%四氧化锇后固定,超薄切片厚50 nm,醋酸铀、柠檬酸铅双染色。置日立Hitach7500透射电子显微镜下观察。
统计学方法 采用SPSS 17.0统计软件对数据进行分析,计量资料以均数±标准差表示,计数资料以百分率表示。两变量相关性分析采用Pearson相关分析,P<0.05为差异有统计学意义。
结 果
一般资料 本组患者多以水肿和尿检异常起病,除2例血压正常外,余10例起病时即血压升高,其中4例存在心脏和大血管病变。全部患者体格检查均未发现指甲和骨骼肌异常(表1)。
实验室检查 本组4例患者确诊时SCr水平升高;7例患者贫血。尿液检查提示本组患者肾小管间质损伤程度较重(表2)。
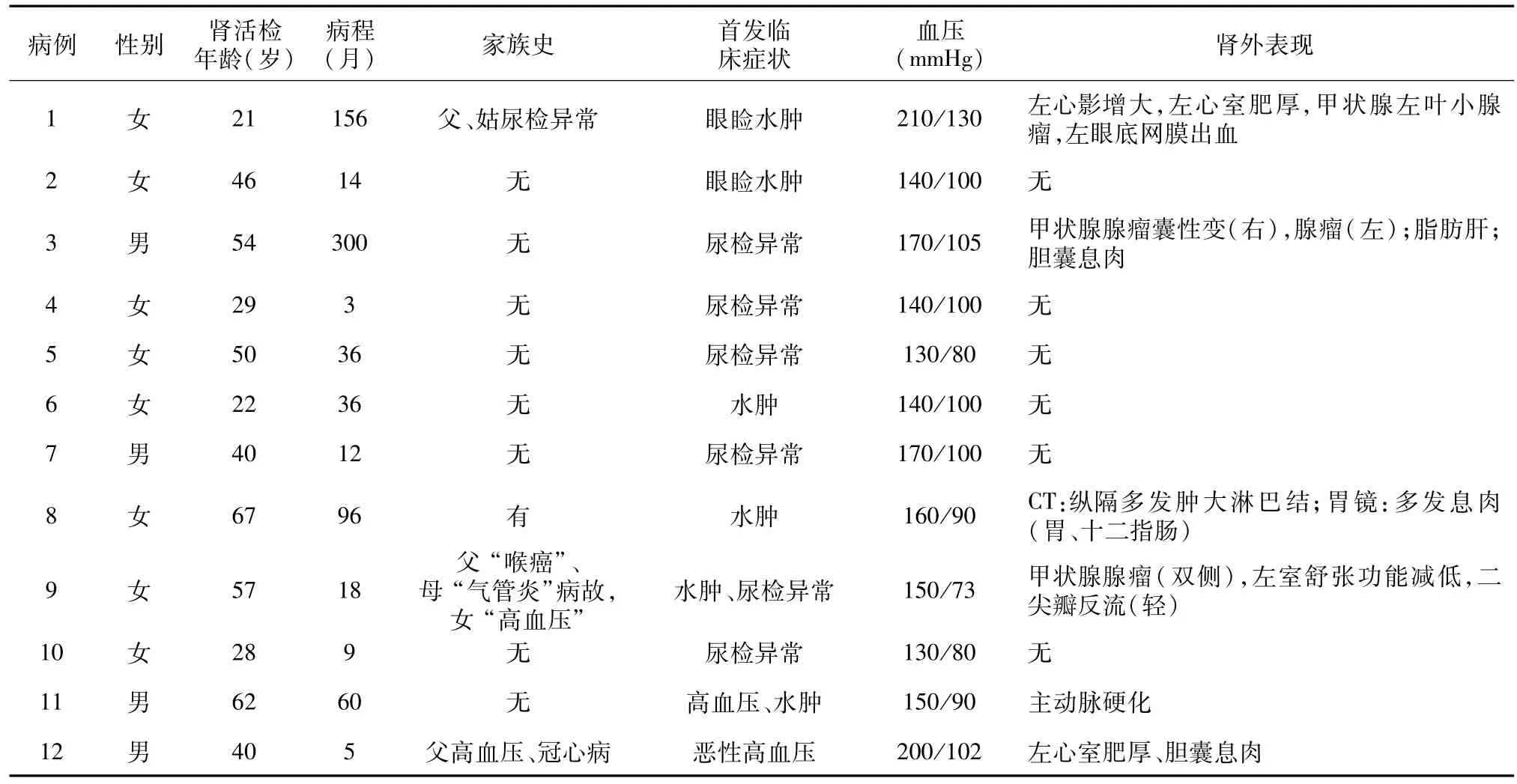
表1 12例胶原Ⅲ肾病患者的一般资料及临床表现
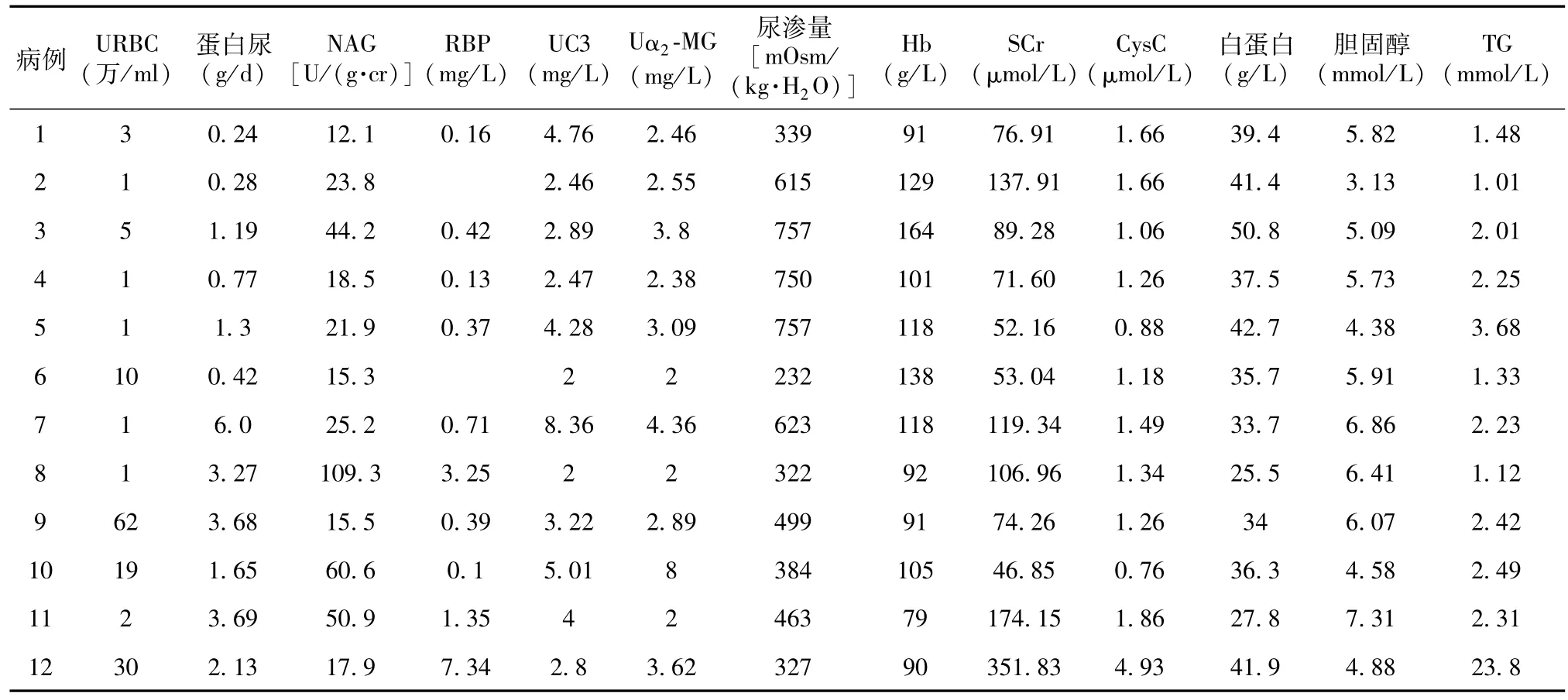
表2 12例胶原Ⅲ肾病患者的实验室检查结果
辅助检查 B超测量双肾体积证实,11例患者双肾体积明显增大(最大者左肾123 mm×43 mm× 54 mm,右肾122 mm×38 mm×44 mm),皮质厚度均增加;仅1例患者双肾体积尚在正常范围内。
肾活检病理特点 组织学观察证实,12例患者肾小球体积均增大,肾小球细胞总数不多,但内皮细胞成对;均存在肾小球内皮下疏松、区域增宽,节段内皮下见均质、嗜伊红淡染物,有时其中混有纤细颗粒状或无定形血浆样物质;节段肾小球毛细血管袢血栓形成,节段内皮下嗜复红物,节段袢与包囊壁黏连;PASM-Masson染色外周袢弥漫“双轨”。我们观察的这组患者肾小球病变大致分为两种类型,一类肾小球呈非增生性“结节样”改变,PAS或PASM-Masson染色肾小球毛细血管袢内皮下被均质、嗜伊红淡染或淡嗜亮绿色的物质充填,致使肾小球毛细血管袢狭小,细胞数减少(系膜细胞和内皮细胞),包囊壁增厚(图1);另类则肾小球“结节样”改变不明显,毛细血管袢腔内内皮细胞成对,PASM-Masson染色外周袢弥漫双轨,但GBM嗜银性弱,呈淡棕色(图2)。肾小管-间质病变与其他肾小球病变类似,包括肾小管萎缩、间质纤维化及淋巴细胞、浆细胞浸润等。
肾间质血管的病变与血压及临床预后直接相关。病例12以恶性高血压起病,间质血管呈TMA样改变,一处入球动脉血栓直接伸入肾小球。尽管患者病程不长,但起病时肾功能已受损,随访时SCr高达708μmol/L。
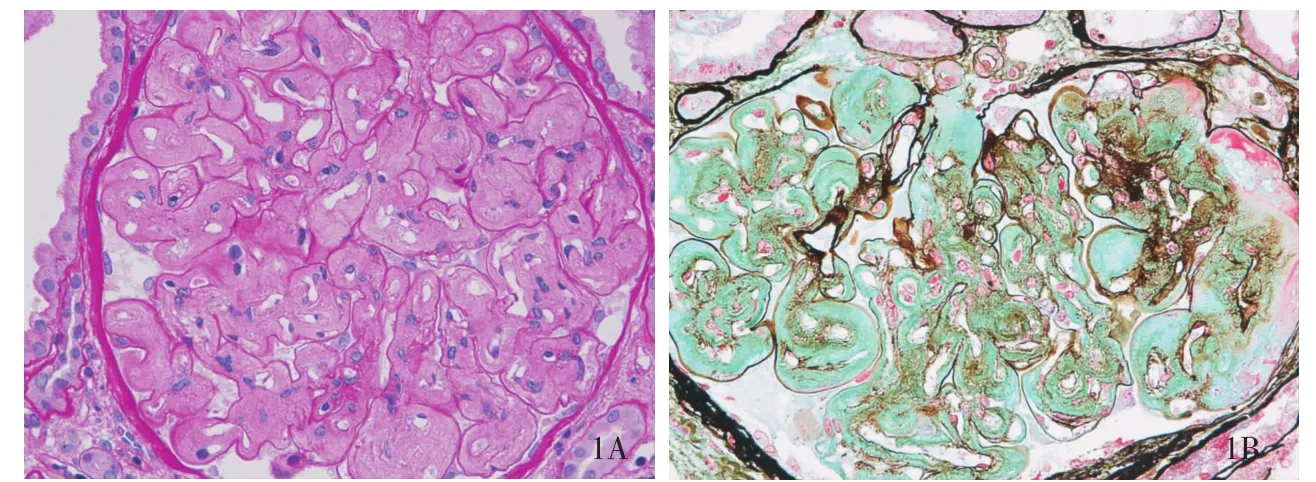
图1 肾小球呈非增生性的“结节样”改变,毛细血管袢内皮下被均质、嗜复红淡染或嗜亮绿淡染的物质充填(A:PAS,× 400;B:PASM-M asson,×400)
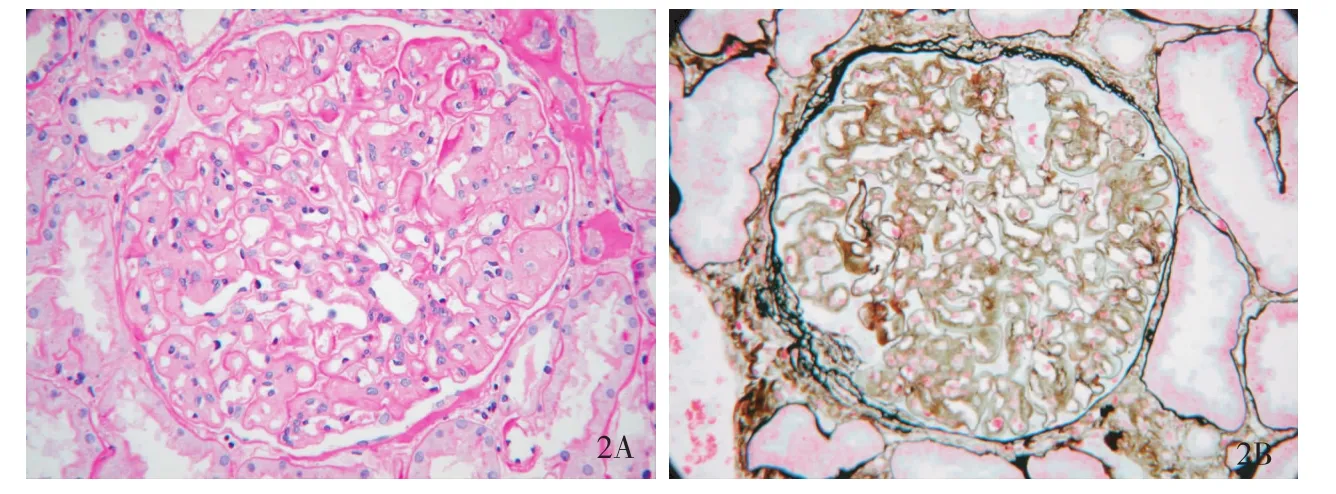
图2 肾小球毛细血管袢内皮细胞成对,外周袢弥漫双轨,(A:PAS,×400;B:PASM-M asson,×400)
全部患者肾组织刚果红染色阴性。
本组患者肾组织免疫荧光观察证实IgG阳性1例,3例IgA,IgM阳性6例,5例C3沉积,1例C4阳性。IgA沉积部位主要在系膜区,IgM沉积无规律,多为非特异性;C3及C4也呈节段分布。
12例患者行石蜡切片胶原Ⅲ染色,4例仅见Ⅲ型胶原沿肾小球毛细血管袢分布,8例除肾小球毛细血管袢阳性外系膜区亦见Ⅲ型胶原阳性(图3)。
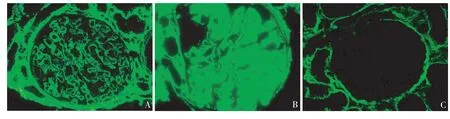
图3 A、B:肾小球毛细血管袢及系膜区胶原Ⅲ染色阳性;C:胶原Ⅲ染色阴性对照(IF,×400)
超微结构观察证实12例患者肾小球毛细血管袢内皮下疏松、区域增宽,内皮下和(或)系膜区见束状或单枝的、有明暗带、直径43~60 nm的胶原纤维(图4);节段肾小球毛细血管袢内皮细胞与GBM剥离,形成透亮区,内皮细胞成对;肾小球足细胞病变包括足突融合、微绒毛化等;偶见壁层上皮细胞节段增生。3例(25%)合并免疫荧光染色IgA阳性者在肾小球系膜区见团块状的电子致密物沉积。
治疗随访 胶原Ⅲ肾病尚缺乏有效治疗方法,本组患者多采用对症治疗,如饮食控制蛋白质摄入,降压(血管紧张素转换酶抑制剂和/或血管紧张素Ⅱ受体拮抗剂),将血压严格控制在130/80mmHg以内。

图4 肾小球毛细血管袢内皮下疏松、区域增宽,内皮下和(或)系膜区见束状或单枝的、有明暗带、直径43~60 nm的胶原纤维(EM)
12例患者平均随访时间20.83±7.86月(范围6~35月),肾活检时SCr升高的4例随访过程中肾功能仍异常,其中病例12出院1月SCr高达708 μmol/L,就诊时肾功能正常者末次随访时1例SCr异常(图5)。肾功能异常者肾活检时血压相对高,但与肾外并发症似乎关系不大。随访末50.0%的患者24h尿蛋白定量<1 g/d;禁水13h尿渗量及尿酶异常者的发生率仍高达66.7%和58.3%。
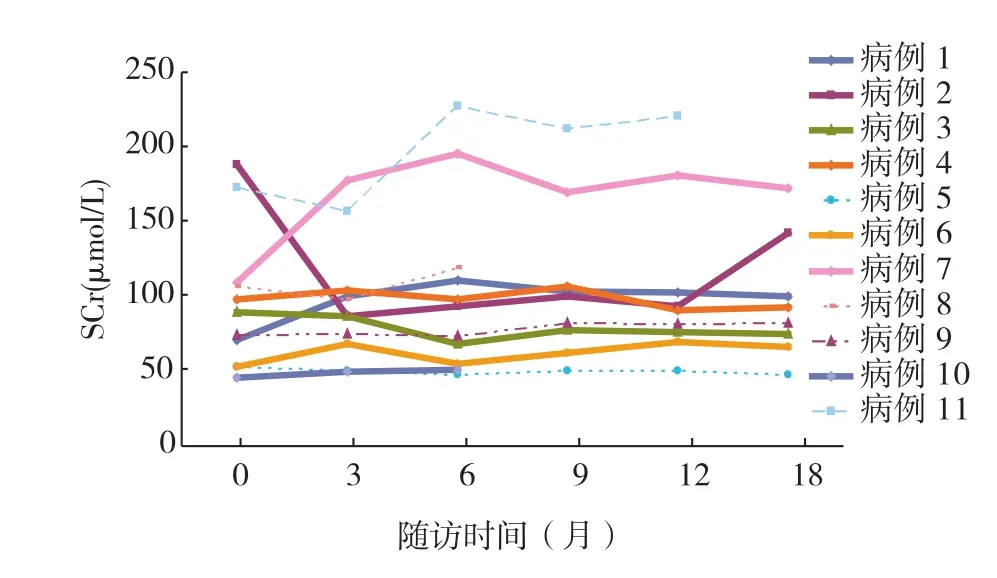
图5 11例患者随访时血清肌酐(SCr)的变化
相关因素分析发现,起病时贫血、24h尿蛋白定量及禁水13h尿渗量与肾功能变化无关,而起病时的收缩压与胱抑素C(CysC)具有相关性,r值= 0.613(P<0.05)(图6)。
讨 论
胶原Ⅲ肾病是较少见的肾小球疾病,本研究统计的4万余例自体肾活检病例中仅12例明确诊断为该病。胶原Ⅲ肾病的诊断要点是电镜观察非硬化GBM内皮下和(或)系膜区、系膜旁区大量胶原沉积,免疫荧光证实其为Ⅲ型胶原[5,6]。
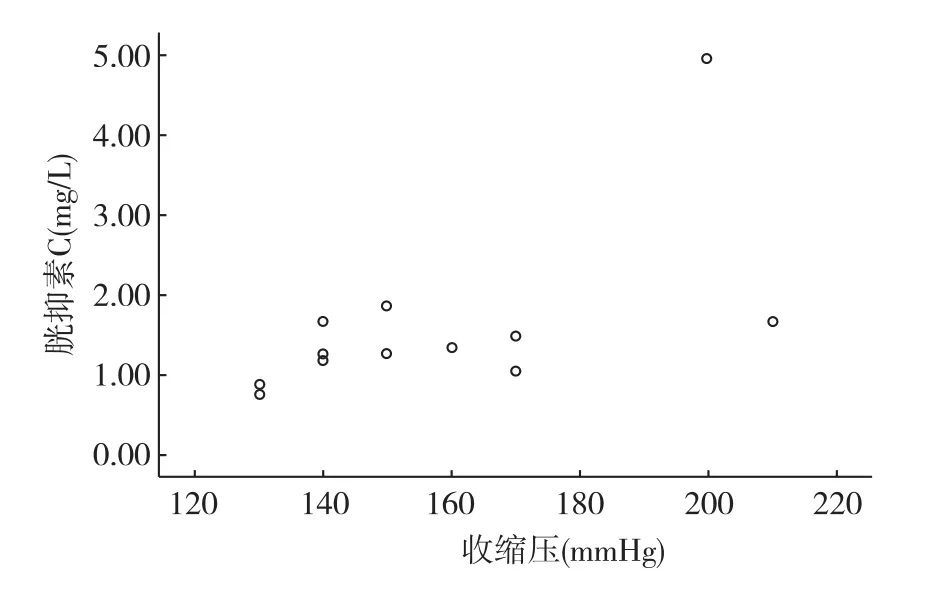
图6 起病时收缩压与胱抑素C的相关性
可能的发病机制 有作者提出,该病为常染色体隐性遗传[7],部分患者可出现遗传性H因子缺乏[8];有的作者则按患者是否存在家族史将其分为家族性和散发性两种[5]。Gubler等[9]报告的10例儿童患者中,7例存在明确的家族史,因此他认为本病可能为常染色体隐性遗传性疾病。Tamura等[10]也报道了两姐妹先后患胶原Ⅲ肾病,因此推测本病可能与遗传相关。Vogt发现本病与遗传性H因子缺乏有关[11],也有作者发现胶原Ⅲ肾病患者肾组织同时存在V型胶原沉积[12]。然而,至今为止胶原Ⅲ肾病确切的发病机制尚不明了。
本组患者仅2例家族成员中存在肾脏病的临床表现。令人遗憾的是,本组患者均未行H因子等检查。因此,其确切的发病机制还有待进一步研究。
临床表现及实验室检查特点 尽管迄今报告的病例中男性多见,但无明确性别差异。发病年龄最小者仅3个月,最大者72岁[13-15]。
Gubler等[9]报告的10例患者发病时年龄均较轻,其中6例患者年龄<2岁,5例幼时即以高血压起病,病程中又有3例血压逐渐增高;早期肾功能多正常或仅有轻度SCr升高,4例患者肾功能进行性减退;随着疾病进展,有的患者3年内即进展为ESRD。肾外症状包括哮喘2例,其中1例死于急性呼吸道危象,2例鱼鳞病(银屑病)。
本组病例多为成年患者,发病时年龄最小者8岁,最大者59岁,无婴幼儿发病者。高血压亦是本组患者最常见、最突出的临床临床表现,10例患者发病时血压即升高,1例8岁起病时发现血压升高(例1)。
本组患者肾外疾病的发生率较高,包括高血压病、冠心病、左心肥厚、甲状腺瘤、胆囊息肉及纵隔多发淋巴结肿大等(表1),有的患者同时合并两种或两种以上肾外症状。少数患者家族成员中见呼吸系统和心血管系统疾病。
本组患者病程迁延,多数在较长时间内肾功能保持稳定,4例患者分别在病程的第14月、12月、60月和第5月出现肾功能减退;这些患者病程中出现脑血栓、哮喘、糖耐量异常和乙肝等并发症。
水肿和蛋白尿是胶原Ⅲ肾病患者最常见的临床表现。蛋白尿可持续存在,也可间歇性发生。尽管本组患者在病程的不同时期均有蛋白尿,但24h尿蛋白定量多寡不一,其中尿蛋白定量<1 g/24h者4例,3例肾病范围的蛋白尿;血尿发生率低,无1例发生肉眼血尿(表2)。
肾小管功能受损是本组患者突出的实验室检查证据,尿RBP升高者4例,尿NAG异常者9例,9例患者低渗尿(表2)。
4例患者肾活检时SCr升高,而CysC异常者8例(66.7%)。与文献报告不十分一致的是,本组患者贫血的发生率较高(7例,58.3%),而Gubler等报告的病例贫血的发生率仅约30%[9]。
肾活检组织学特征 多数作者认为胶原Ⅲ肾病肾组织改变无特殊性,除肾小球体积增大,肾小球基膜弥漫分层外,一般肾小球系膜细胞无增生,特染(PAS和/或Masson三色)可见GBM内和系膜区弱嗜复红或嗜亮绿的物质沉积,致使毛细血管袢腔受挤压甚至闭塞。少数病例可见节段内皮下沉积物,但未见上皮侧沉积。疾病早期小管-间质及血管无异常。亦有报道少数病例存在明显的小管-间质改变,小动脉内皮下见透明沉积物。疾病晚期重复活检时肾小球硬化可达60%~80%,同时伴严重的肾小管和间质病变[9,14-17]。
我们认为,胶原Ⅲ肾病患者肾小球毛细血管袢内皮细胞病变是其基础组织学改变,由于肾小球内皮细胞成对,弥漫内皮下疏松区域增宽,PASMMasson染色时见肾小球外周袢分层。从观察的病变见一类肾小球分叶不明显,细胞总数正常或增多(内皮细胞数增多为主),节段系膜区增宽外周袢“分层”;而另一类则肾小球呈非增生性“结节样”病变,肾小球轻分叶,肾小球在不同染色(HE、PAS、Masson三色)切片均呈均质(嗜伊红、嗜复红、嗜亮绿)淡染物挤压毛细血管袢腔致使袢狭窄,其中可见小或粗颗粒或纤细的物质,偶见红细胞,节段内皮下渗出性病变;肾小球细胞数减少(无论内皮细胞还是系膜细胞),见系膜溶解。这两种组织学病变患者的肾小球均可见毛细血管袢节段塌陷,形成塌陷区,袢与囊壁黏连也无差别。肾小管间质慢性病变也与肾小球改变无关联。肾间质血管透明变性十分常见,但与血压高低无相关性。
我们尚未找出两种肾小球组织病变与病程、性别、年龄的关系,文献也未提及这样的改变是否与肾功能相关。但对照本组3例呈非增生性结节样病变者肾功能的状况,证实2例肾活检时肾功能减退,1例随访至病程102月时SCr轻度升高。而以肾小球内皮细胞病变为主要病变的患者,肾活检时年龄21~67岁,2例分别在病程的14月和5月SCr升高,其余患者病程长短不一(表1),即使病程已达156月或300月,肾活检及随访时肾功能亦无异常。
至今,无论国外还是国内文献至今尚未提及胶原Ⅲ肾病是否存在这两种不同的组织类型,也未证实肾功能变化与肾活检病理类型有一定关系,更不清楚这两种病变是疾病的不同阶段还是存在不同的发病机制,他们是否与疾病进展有关联。
电镜下可见不同程度的系膜区增宽、基质增加,内皮下间隙增宽,在增宽的系膜区、系膜旁区及内皮下区域。肾小球见电子致密的、散在或束状聚集的纤维状物积聚,纤维有明、暗带,多有周期性横纹间隔,直径多在40~70 nm间。致密层未见纤维沉积。即使电镜观察证实存在这些病变,也必须行免疫荧光染色确定这些胶原纤维是Ⅲ型胶原。
肾小球系膜区增宽,但系膜细胞不增生,一些袢可见系膜基质插入至内皮下,并见新形成的基膜(光学显微镜下的双轨)。足细胞足突局灶/弥漫融合,膜上无电子致密物沉积;一些病例可见节段内皮下和系膜区电子致密物。内皮细胞肿胀,富胞质,胞质连拱状。包囊壁、小动脉和肾小管基膜无明确病变[9,14-17]。
本组病例中3例肾组织免疫荧光染色证实肾小球系膜区有IgA沉积,电镜观察亦见肾小球系膜区团块状的电子致密物,从而证实胶原Ⅲ肾病合并IgAN。
诊断及鉴别诊断 现已明确组成GBM的主要成分是Ⅳ型胶原,肾小球中也可见很少的Ⅴ型和Ⅵ型胶原,但它们仅局限在内皮下间隙和系膜区[15,18]。有人提出胶原Ⅲ肾病的发生可能与胶原代谢异常有关,在部分患者中也证实血清胶原Ⅲ前肽水平升高。因此认为检测血清胶原Ⅲ前肽可作为诊断胶原Ⅲ肾病的非创伤性指标[12,14],但至今公认的肾活检病理检查仍是诊断本病的唯一手段。
正常人类GBM和系膜基质中无Ⅲ型胶原,然而,在约1/3的某些肾脏疾病如IgA肾病、膜增生性肾小球肾炎、新月体肾炎及硬化性肾炎患者中系膜区经免疫荧光证实可见Ⅲ型胶原。多数人认为这种疾病状态下的Ⅲ型胶原起源于系膜细胞,也不除外肾小球外产生的可能性。然而此种状态下Ⅲ型胶原分布的特点是很局限,呈节段性分布,且仅在受检组织0.33%的肾小球中检出[15,18]。
根据组织学、免疫病理及超微病理检查,本病诊断并不困难,但诊断时要注意行免疫荧光胶原Ⅲ染色时必须有阳性和阴性对照组,电镜应避免观察硬化部位,因为硬化处可见胶原纤维[19]。
此外,胶原Ⅲ肾病还需与以下疾病鉴别:(1)甲-髌综合征:临床除存在指甲-髌骨发育异常外,电子显微镜下常见基膜不规则增厚,伴明显的透亮区,异常的胶原纤维见于致密层和内疏松层,系膜区少见且无系膜基质插入至内皮下;相反胶原Ⅲ肾病患者无指甲-髌骨发育异常,系膜区、内皮下(内疏松层)增宽[19-21],且见胶原纤维;(2)膜增生性肾炎:其特点为高度增生的肾脏固有细胞,系膜区增宽基质增多,并插入至内皮下,系膜细胞增生明显,毛细血管袢内皮下较多电子致密物,免疫荧光检查也有助鉴别[19,20];(3)血栓性微血管病:尽管血栓性微血管病患者光镜下肾小球外周袢内皮下区域增宽与胶原Ⅲ肾病相似,但电镜观察前者在增宽的内皮下区域中可见无定形的物质、细胞碎屑和变形的红细胞,内皮细胞与基膜剥离,无系膜区胶原沉积,而后者增宽的内皮下区域中则以有明、暗带的束状、蠕虫状或逗号样的、直径约40~70 nm的胶原沉积为主,临床表现也有助两者鉴别[19,20];(4)纤维性肾小球病(fibrillary glomerulopathy,FGP)以肾小球内非刚果红染色的微细纤维沉积为特征,与本病的主要区别在于FGP常常为免疫介导的疾病,组织学改变以膜性、膜增生性及系膜病变为特征。电子显微镜观察两病在肾小球中的纤维成分不一,FGP者纤维直径约20 nm,排列杂乱,无固定位置,而本病为成束、粗大的有明、暗带的胶原纤维,直径约为40~70 nm,且有特定的沉积部位[18,19];(5)纤维连接蛋白肾小球病(fibronectin glomerulopathy,FNG)是新发现的以纤维样物质沉积的另一种肾小球疾病,临床上多见肾脏范围的蛋白尿,组织学以肾小球系膜增宽,电镜观察GBM增厚为特征,可见直径10 nm的纤维样物质沉积,免疫组织化学染色证实肾小球内存在纤维连接蛋白[19-21];(6)免疫管状肾小球病(immunotactoid glomerulopathy,ITG)是另一种不常见的在肾小球中可见纤维样物质沉积的疾病。纤维的特点是中空或圆柱状的,直径30~40 nm,光镜下以系膜病变为主,免疫荧光染色粗颗粒的IgG和C3在系膜区或外周袢分布。患者以蛋白尿、肾病综合征或血尿为主要临床表现,ITG常与潜在的淋巴增生紊乱有关[18,19]。
本病尚无有效的治疗方法,多数患者肾功能呈缓慢减退。本组病例中有的病史已达25年,而SCr仍正常。相关分析发现,血压与CysC水平相关,因此积极控制血压,可能对延缓肾功能的进展具有重要意义。
1 Alchi B,Nishi S,Narita I,et al.Collagenofibrotic glomerulopathy:clinicopathologic overview of a rare glomerular disease.Am JKidneyDis,2007,49(4):499-506.
2 Ikeda K,Yokoyama H,Tomosugi N,etal.Primary glomerular fibrosis:a new nephropathy caused by diffuse intra-glomerular increase in atypical type III collagen fibers.Clin Nephrol,1990,33(4):155 -159.
3 Churg J,Bernstern J,Glassok RJ.Collagenofibrotic glomerulopathy// Churg J(ed).Renal disease classification and atlas of glomerular disease(ed 2).New York,NY,Igakushoin,1995,chapt 17,P406 -443.
4 Ferreira RD,Custódio FB,Guimarães CS,et al.Collagenofibrotic glomerulopathy:three case reports in Brazil.Diagn Pathol,2009,4:33.
5 Abt AB,Cohen AH.Newer glomerular disease.Semin Nephrol,1996,16(6):501-510.
6 Yoshida F,Yuzawa Y,Shigematsu H,et al.Nephrotic syndrome with massive accumulation of type I andⅢcollagen in the glumeruli. Intern Med,1993,32(2):171-176.
7 Kim Y,Vernier RL,Fish AJ,et al.Immunofluorescence studies of dense deposit disease.The presence of railroad tracks and mesangial rings.Lab Invest,1979,40(4):474-480.
8 Pangburn MK,Schreiber RD,Muller-Eberhard HJ.Human complement C3b inactivator:isolation,characterization,and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution.JExp Med,1977,146(1):257-270.
9 Gubler MC,Dommergues JP,Foulard M,et al.Collagen typeⅢglomerulopathy:a new type of hereditary nephropathy.Pediatr Nephrol,1993,7(4):354-360.
10 Tamura H,Matsuda A,Kidoguchi N,et al.A family with two sister with collagenofibrotic glomerulonephopathy.Am JKidney Dis,1996,27(4):588-595.
11 Vogt BA,Wyatt RJ,Burke BA,etal.Inherited factor H deficiency and collagen typeⅢglomerulopathy.Pediatr Nephrol,1995,9(1):11 -15.
12 Morita H,Hasegawa T,Minamoto T,et al.Collagenofibrotic glomerulopathy with a widespread expression of type-V collagen. Virchows Arch,2003,442(2):163-168.
13 Gubler MC,Dommergues JP,Furioli J,et al.Syndrome de“Nail-Patella”sans atteinte extrarenale.Une nouvelle nephropathie hereditaire glomerulaire.Ann Pediatr(Paris),1990,37(2):78-82.
14 Mizuiri S,Hasegawa A,Kikuchi A,et al.A case of collagenofibrotic glomerulopathy associated with hepatic perisinusoidal fibrosis. Nephron,1993,63(2):183-187.
15 Yoshioka K,Takemura T,Tohad M,et al.Glomerular localization of typeⅢcollagen in human kidney disease.Kidney Int,1989,35(5):1203-1211.
16 Ikeda K,Yokoyama H,Tomosugi N,etal.Primary glomerular fibrosis:a new nephropathy caused by diffuse intraglomerular increase in atypical typeⅢcollagen fibers.Clin Nephrol,1990,33(4):155 -159.
17 Imbasciati E,Gherardi G,Morozumi K,et al.Collagen typeⅢglomerulopathy:a new idiopathic glomerular disease.Am JNephrol,1991,11(5):422-429.
18 Morel-Maroger Striker L,Killen PD,Chi E,et al.The composition of glomerulosclerosis.I.Studies in focal sclerosis,crescentic glomerulonephritis,andmembranoproliferative glomerulonephritis.Lab Invest,1984,51(2):181-192.
19 Herrera GA,Turbat-Herrera EA.Renal diseases with organized deposits:an algorithmic approach to classification and clinicopathologic diagnosis.Arch Pathol Lab Med,2010,134(4):512 -531.
20 Gohen AH,Addler SG.Nail-patella syndrome//Tisher CC,Brenner BM(eds).Renal pathology.Lipponcott,Philadelphia,1989:P1191-1195.
21 Tagushi T,Takebayashi S,Nishimura M,et al.Nephropathy of nailpatella syndrome.Ultrastruct Pathol,1988,12(2):175-183.
Collagen typeⅢglomerulopathy:M orphological features and clinicalmanifestations
CHEN Hui-ping,XU Feng,HUANGQian,HUANGGao-yuan,XIEHong-lang,ZHUMao-yan,ZHANGMing-chao,HEQian,LIANG Shao-shan,ZENGCai-hong,LIU Zhi-hong
Research Institute of Nephrology,Jingling Hospital,Nanjing University Clinical School of Medicine,Nanjing 210002,China
Objective:Collagen typeⅢglomerulopathy is a non-immune-mediated glomerular disease,and also a form of glomerulopathy in which organized collagen typeⅢprogressively deposits.We analyzed the renal morphological features and laboratory examinations,as well as clinicalmanifestations,in patients with collagen typeⅢglomerulopathy to improve diagnostic level of this disease. M ethodology:Twelve caseswith collagen typeⅢglomerulopathy proved by light microscopy,immunofluorescence and electron microscopy were retrospectively studied.Their clinical materials,laboratory findings and pathological data were collected. Results:There were 4 males and 8 females aged from 21 to 67 years old. Themean duration of disease was(62.1±60.9)months(ranged from 3 to 300 months).Proteinuria and hypertension were themost common onset clinical symptomswith the incidence both of83.3%.Most cases had normal renal function at making diagnosis except four cases(33.3%).The increase of red blood cell in urinary sedimentwas presented in only one case(8.3%).Most cases had tubular injury including elevated urine NAG in 9(75.0%),increased urine RBP in 4 and decreased urine osmolality in all cases.Immunofluorescence microscopy revealed that collagen typeⅢwas abundant staining in the non-sclerosis glomerular capillary loops and/ormesangium,and C3 and IgA were positive in 5(41.7%)
and 3(25.0%)cases,respectively.In the lightmicroscopy,glomerular tuftswere globally expanded and the subendothelial spaces of glomerular basementmembrane weremarkedly expanded in all cases.There were two typic patterns in glomerular changes.One was that the globular deposition of eosinophilic substances in the expanded mesangial area was observed,narrowing of capillary lumen in glomerulus and a decrease in nuclearity of themesangial area and endothelium was noted,and Bowman's capsuleswere thickened.Another was themarked enlargementof the glomerular tuft due to both expansion of themesangialmatrix and thickening of the capillary walls.On PAS and silver-stained,the thickened capillary walls had a double-contour appearance.Electron microscopy showed that all cases had expanded subendothelial space of glomerular basementmembrane giving a lucent or lytic appearance to these structures.The marked accumulation of fibrillar material with bunchy or single and with a transverse band structure could be identified in subendothelial space and mesangium.The diameters of these fiberswere 43~60 nm.3 caseswho had positive IgA staining in immunofluorescence had electron-dense deposits presented in mesangium.During follow-up,the SCr was elevated still in 4 cases(33.3%). Conclusion:Hypertension and proteinuria are the most common clinical features of collagen typeⅢglomerulopathy.Morphological Changes are characteristic,while immunofluorescence microscopy and electron microscopy are imperative to establish a definitive diagnosis.
collagen typeⅢglomerulopathy renal biopsy hypertension proteinuria
2011-01-03
(本文编辑 律 舟)
南京军区南京总医院全军肾脏病研究所(南京,210002)
©2011年版权归《肾脏病与透析肾移植杂志》编辑部所有
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
天津医科大学学报(2021年4期)2021-08-21
肾脏病与透析肾移植杂志(2017年2期)2017-05-09
临床与实验病理学杂志(2017年12期)2017-03-20
安徽医科大学学报(2016年12期)2017-01-15
医学研究杂志(2015年9期)2015-07-01
肾脏病与透析肾移植杂志(2015年5期)2015-06-09
中国实验诊断学(2015年10期)2015-05-06
中华皮肤科杂志(2014年3期)2014-12-19
中国火炬(2014年8期)2014-07-24
