
青霉胺联合硫普罗宁治疗肝型肝豆状核变性的临床研究
2011-06-02 03:11徐磊蔡永亮王艳昕侯志峰许珍晶徐明安马守亮
中国神经精神疾病杂志 2011年7期
徐磊 蔡永亮 王艳昕 侯志峰 许珍晶 徐明安 马守亮
肝豆状核变性(hepatolenticular degeneration,HLD)是一种常染色体隐性遗传的铜代谢障碍疾病,临床上主要表现为进行性加重的锥体外系症状、肝硬化、肾功能损害及角膜 Kayser-Fleischer(K-F)环形成等[1]。肝型HLD则以持续性血清转氨酶增高、急性或慢性肝炎、肝硬化或暴发性肝功能衰竭等肝病症状为主要临床表现[2]。目前治疗HLD的药物主要为重金属络合剂,首选药物为青霉胺(penicillamine,PCA),其将铜络合后从尿液排出以减少铜在体内蓄积。硫普罗宁(tio-pronin,TPO)是一种新型的含巯基甘氨酸类药物,其化学名为N-(2-巯基丙酰基)甘氨酸,除具有改善肝功能的作用外,又具有络合重金属的作用[3]。本实验运用PCA联合TPO治疗肝型HLD,并观察其临床疗效和安全性。
1 资料与方法
1.1 一般资料 选择于2008年1月至2010年8月入住我科年龄大于12岁的肝型HLD患者33例,诊断标准参考肝豆状核变性的诊断和治疗指南[2],入选病例均符合指南诊断标准且排除病毒性肝炎、自身免疫性肝病等其他肝病。所有病例在入科前未接受正规驱铜治疗,排除青霉胺或硫普罗宁过敏者。33例患者按不平衡指数最小分配原则随机分为对照组和治疗组。对照组16例,其中男10例,女6例,年龄12岁至26岁,平均(17.1±4.5)岁。病程1个月至36个月,平均(10.9±9.5)个月。治疗组17例,男9例,女8例,年龄12岁至24岁,平均(16.4±3.4)岁。病程1个月至30个月,平均(12.2±7.8)个月。
1.2方法两组予以低铜饮食和锌剂(150 mg/d)口服,在此基础上对照组给予PCA(上海医药集团有限公司信谊制药总厂生产,国药准字:H31022286)250 mg口服,每日3次;治疗组给予PCA 250 mg口服,每日3次,同时加TPO(河南省新谊药业股份有限公司,国药准字:H41020799)200 mg口服,每日3次。疗程均为12周。于用药前1 d和治疗结束后1 d检测24 h尿铜(urinary copper,u-Cu)、血清铜(plasma copper,p-Cu)和铜蓝蛋白(ceruloplasmin,CER)水平,估算游离铜(nonceruloplasmin plasma copper,nCER-Cu)水平,检测天门冬氨酸氨基转移酶(aspartate aminotransferase,AST)、总胆红素(total bilirubin,TBIL)、白蛋白(albumin,ALB)、国际标准化比值(international normalized ratio,INR)以及白细胞(white blood cell,WBC)、血小板(blood platelets count,BPC)水平,观察有无不良反应。nCER-Cu水平(μg/dL)数值上 =p-Cu(μg/dL)- CER(mg/dL)×3[4]。正常范围为 10 ~15 μg/dL[5]。计算 Dhawan et al[6]指数评估肝功能损害严重程度和预后。
1.3 统计学方法
采用SPSS17.0对资料进行分析,对各组数据先采用Kolmogorov-Smirnov检验法进行正态性检验,若符合正态性分布,则进行独立样本t检验,数据以±s形式表示;若不符合正态分布,则进行Mann-Whitney检验。检验水准α=0.05。
2 结果
2.1 基线资料 两组患者的年龄、性别、病程及肝功能损害情况均无统计学差异(P>0.05),基线资料具有可比性(表1)。
2.2 治疗前后u-Cu和nCER-Cu水平 两组u-Cu水平均较治疗前显著升高(P<0.01),与对照组比较,治疗组升高更为显著(P<0.05);两组nCER-Cu水平较治疗前显著降低(P<0.05),与对照组比较,治疗组降低更为显著(P<0.05)(表2)。
2.3 治疗前后肝功能、血常规和Dhawan指数 治疗组AST和TBIL水平较治疗前及对照组显著下降(P<0.01),而对照组无显著变化(P>0.05),两组治疗后WBC、BPC、ALB和INR水平较治疗前及组间比较均无统计学差异(P>0.05)。治疗组Dhawan指数较治疗前显著改善(P<0.01),与对照组无统计学差异(P>0.05),而对照组较治疗前无显著变化(P>0.05)(表3)。
2.4 不良反应 对照组1例和治疗组2例患者随访中拒绝继续参加实验。共30例患者完成为期12周的治疗。30例患者中有8例(26.7%)出现不同程度的不良反应,如白细胞和血小板减少(对照组2例、治疗组1例)、胃肠道反应(对照组2例、治疗组3例)、皮疹(两组均为2例)。不良反应均在短暂停药或对症治疗后缓解。
3 讨论
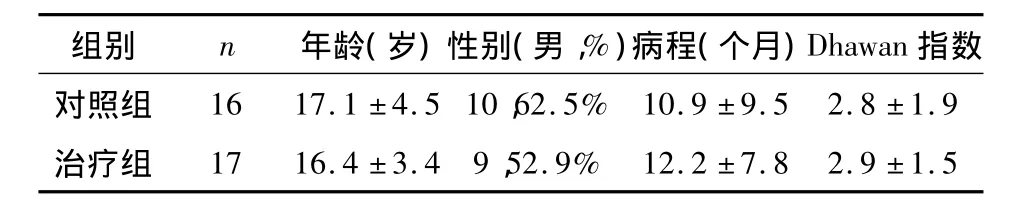
表1 基线资料

表2 治疗前后u-Cu与nCER-Cu水平
HLD是少数几种可以治疗的常染色体隐性遗传性疾病之一[7]。目前治疗HLD的药物有两大类,一类为络合剂,能强力促进铜离子排出,另一类为是阻止肠道对外源性铜的吸收。络合剂中PCA疗效肯定、药源充足、价格低廉、使用方便,目前在我国仍作为治疗HLD的主要药物[2]。TPO是一种与PCA性质相似的含巯基药物,能通过提供巯基络合重金属,能防止四氯化碳、乙硫氨酸、对乙酰氨基酚等造成的肝脏损害,还可以通过巯基与自由基的可逆结合,清除自由基[8-9]。我们既往运用PCA联合TPO治疗铜负荷大鼠,能够明显提高驱铜效果以及改善肝功能[10]。本研究中两者联合治疗肝型HLD患者,同样疗效满意。
表3 两组患者治疗前后肝功能、血常规和Dhawan指数比较(±s)

表3 两组患者治疗前后肝功能、血常规和Dhawan指数比较(±s)
Dhawan index对照组 治疗前 16 2.12±0.95 110.6±39.3 3.69±0.75 1.37±0.36组别 n TBIL(mg/dL)AST(U/L)ALB(g/dL)INR WBC(109/L)BPC(109/L)4.7±1.7 171.4±71.4 2.8±1.9治疗后 15 1.71±0.53 88.5±25.8 3.77±0.57 1.27±0.27 4.6±1.4 155.6±54.6 2.0±1.2治疗组 治疗前 17 2.17±1.09 119±41.0 3.77±0.64 1.35±0.38 4.3±1.9 144.4±65.0 2.9±1.5治疗后 15 1.28±0.35 52.9±19.6 3.85±0.55 1.26±0.23 4.5±1.6 135.8±50.6 1.5±0.9
24 h尿铜是HLD驱铜疗效评判的客观指标,能够反映循环中未与铜蓝蛋白结合的铜水平[7]。在本实验中,对照组和治疗组都表现出良好的驱铜效果,24 h尿铜水平均显著高于治疗前(P<0.05),但是治疗组表现出更佳的疗效(P<0.05),表明PCA和TPO具有协同驱铜作用,要优于单用PCA。游离铜水平反映可自由沉积于组织的铜水平,主要反映铜毒性,游离铜很难测量,但是可以通过血清铜和铜蓝蛋白水平估算,其正常范围为10~15 μg/dL[7]。在本实验中,两组游离铜水平均较治疗前均有显著下降(P<0.05),但是治疗组的游离铜水平降低更显著、更接近正常范围(P<0.05),说明在相同的疗程内PCA和TPO联用能更好地降低游离铜水平,更有效地降低游离铜的毒性。
另外,本实验中治疗组AST和TBIL水平较治疗前显著下降(P<0.05),明显优于对照组(P<0.05)。TPO具有明确的改善肝功能作用[11]。对照组的AST和TBIL水平与治疗前比较无统计学差异(P>0.05),但是水平也有下降,说明驱铜时减少肝铜负荷能够协助改善肝功能。既往文献表明[12-13],单用青霉胺治疗HLD,其肝功能全面改善需要数年的时间,本实验随访未达到如此长的时间。我们认为肝型的HLD患者在驱铜的同时,还需要正规持续的保肝治疗,可加速肝功能的改善。在本实验中,两组均未能明显改善HLD患者WBC和BPC水平。HLD患者部分合并脾脏大、脾功能亢进,引起WBC和BPC水平下降,临床上多采取脾切除术来改善过低的WBC和BPC水平,而药物治疗效果微乎其微。本实验中两组患者的ALB和INR水平无显著变化。治疗组Dhawan指数较治疗前显著改善(P<0.05),对照组Dhawan指数较治疗前比较虽然无统计学差异(P=0.159),但是指数还是有所下降。无论是对照组还是治疗组,患者的肝功能均未见到进一步恶化的趋势。
本实验证实,PCA联合TPO比单独运用PCA疗效更佳,具有良好的驱铜和改善肝功能效果。本实验还表明二者联合用药是安全的,较单用青霉胺相比未见毒副反应风险的增加。此外,本实验中需进一步扩大样本量及延长观察时间,我们将在以后的实验中进一步完善。
[1]Ala A,Walker AP,Ashkan K,et al.Wilson's disease[J].Lancet,2007,369(9559):397 - 408.
[2]中华医学会神经病学分会帕金森病及运动障碍学组,中华医学会神经病学分神经遗传病学组.肝豆状核变性的诊断和治疗指南[J].中华神经科杂志,2008,41(8):566-569.
[3]Dahl JA,Maddux BL,Hutchison JE.Toward Greener Nanosynthesis[J].Chem Rev,2007,107(6):2228 - 2269.
[4]Brewer GJ,Askari F,Lorincz MT,et al.Treatment of Wilson disease with ammonium tetrathiomolybdate:IV.Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease[J].Arch Neurol,2006,63(4):521 -527.
[5]Pfeiffer RF.Wilson’s Disease[J].Semin Neurol,2007,27(2):123-132.
[6]Dhawan A,Taylor RM,Cheeseman P,et al.Wilson's disease in children:37-year experience and revised King's score for liver transplantation[J].Liver Transpl,2005,11(4):441 - 448.
[7]Roberts EA,Schilsky ML.Diagnosis and Treatment of Wilson Disease:An Update[J].Hepatology,2008,47(6):2089 -2111.
[8]Garcia MS,Sanchez-Pedreño C,Albero MI,et al.Determination of penicillamine or tiopronin in pharmaceutical preparations by flow injection analysis[J].J Pharm Biomed Anal,1993,11(8):633 -638.
[9]Domingo JL.Prevention by chelating agents of metal-induced developmental toxicity[J].Reprod Toxicol,1995,9(2):105 - 113.
[10]徐磊,蔡永亮,徐志树,等.凯西莱联合青霉胺治疗肝豆状核变性模型鼠的研究[J].临床神经病学杂志,2007,20(6):450-452.
[11]付玉军,田恩照,吴某来,等.硫普罗宁注射液干预大鼠肝星状细胞致肝纤维化形成作用的研究[J].中国医师杂志.2007,9(7):901-903.
[12]Shiono Y,Wakusawa S,Hayashi H,et al.Iron accumulation in the liver of male patients with Wilson’s disease[J].Am J Gastroenterol,2001,96(11):3147 -3151.
[13]Grand RJ,Vawter GF.Juvenile Wilson disease:histologic and functional studies during penicillamine therapy[J].J Pediatr,1975,87(6 Pt 2):1161 -1170.
猜你喜欢
广州化学(2022年4期)2022-09-01
传染病信息(2022年2期)2022-07-15
现代临床医学(2022年3期)2022-06-06
现代临床医学(2021年4期)2021-07-31
中国金属通报(2021年20期)2021-03-11
生物技术通报(2021年12期)2021-02-10
商品与质量(2019年31期)2019-11-28
天津医科大学学报(2019年3期)2019-08-13
中兽医学杂志(2019年1期)2019-01-06
中国当代医药(2015年26期)2015-03-01
