
BRCA1基因甲基化及甲硫氨酸合成酶与乳腺癌发病的关系
2011-05-30 01:30马文慧侯琳韩琳琳
中国癌症杂志 2011年4期
马文慧 侯琳 韩琳琳
青岛大学医学院生物化学与分子生物学教研室,山东 青岛 266021
D NA甲基化是肿瘤发生的一个早期特征,通过基因组总体水平低甲基化和某些启动子区域高甲基化造成基因功能的失活,从而导致肿瘤的形成[1]。S-腺苷甲硫氨酸(S-adenosyl-L-methionine,SAM)是体内甲基的直接供体,通过甲硫氨酸循环及叶酸代谢得到补充,故推测此循环障碍可能导致甲基化异常。甲硫氨酸合成酶(methionine synthase,MS)是参与甲基供体生成的关键酶,已发现MS基因多态性与乳腺癌发病相关[2],其多态性可能通过影响基因的表达,使酶水平或活性改变,导致DNA合成紊乱和甲基化异常[3]。BRCA1基因是非常重要的乳腺癌易感基因和抑癌基因,散发性乳腺癌罕见BRCA1基因突变,但癌组织中该基因表达却显著下降,目前研究认为这与BRCA1启动子甲基化有关,但对BRCA1基 因甲基化机制的报道尚显不足。散发性乳腺癌BRCA1甲基化与肿瘤分级[4]、癌组织分型[5]、激素受体[6-7]相关,但其与雌激素受体(estrogen receptor,ER)阴性/阳性相关尚无定论。本研究首先检测乳腺癌组织及相应癌旁组织、乳腺良性病变组织中BRCA1 mRNA的表达水平及甲基化状态,同时检测叶酸代谢过程中关键酶MS基因的表达,探讨该酶与BRCA1基因甲基化及其与乳腺癌发生的关系,从而为肿瘤甲基化机制及乳腺癌的发病机制提供新的理论依据。
1 资料和方法
1.1 资料
1.1.1 乳腺癌组织标本
病例组选取2009年8月—2009年11月在青岛大学医学院附属医院行外科 手术切除新鲜组织标本71例,其中良性病变组织9例、乳腺癌及癌旁组织(距癌>3 cm)各31例(获知情同意),标本切除后迅速放入-70 ℃冰箱保存。术前所有患者皆未接受治疗,术后均经病理组织学确诊。全部乳腺癌病例户籍均为山东地区,无家族乳腺癌发病史。
1.1.2 主要试剂
MMLV逆转录试剂盒和Wizard DNA clean up纯化试 剂盒购自美国Promega公司,对苯二酚和亚硫酸氢钠购自美国Sigma公司,耐热DNA
聚合酶购自大连宝生物公司,饱和酚购自北京索莱宝科技有限公司。
1.1.3 主要仪器
GeneAmp2400PCR反应仪(PE公司),紫外分光光度计(Eppendorf公司),5415D高速离心机(Eppendorf公司),5417R高速冷冻离心机(Eppendorf公司)
1.2 方法
1.2.1 组织DNA、RNA提取
采用酚-氯仿抽提法提取组织DNA,DNA经紫外分光光度计定量,-20 ℃保存备用。参照FASTAGEN-RNAfast200试剂盒(上海飞捷生物技术公司产品)操作说明提取组织总RNA。RNA经琼脂糖凝胶电泳鉴定其完整性,通过紫外分光光度计测定D260(260 nm光密度值)与D280(280 nm光密度值),计算D260/D280比值,计算RNA的纯度与浓度。用MMLV逆转录酶进行逆转录反应,合成cDNA。
1.2.2BRCA1基因的反转录PCR (RT-PCR)
用2 μg总RNA进行逆转录。cDNA经半定量PCR进行扩增,BRCA1及内参照β-actin特异性引物的序列和扩增片段长度见表1。PCR为25 μL反应体系,反应条件: 94 ℃预变性5 min;94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,30个循环;72 ℃终延伸7 min。PCR产物在含溴化乙锭的2%琼脂糖凝胶中电泳,由Quantity One 4.6.6 (BIO-RAD)分析条带D值。
1.2.3 组织DNA的亚硫酸氢盐处理
在50 μL反应体系中加入2 μg DNA和3 mol/L NaOH 5.5 μL,42 ℃水浴30 min,再加入新鲜配制的对苯二酚30 μL,3.6 mol/L亚硫酸氢钠520 μL,50 ℃水浴16 h。采用Wizard DNA clean-up system (Promega公司)纯化试剂盒纯化修饰过的DNA。纯化后的DNA加入3 mol/L NaOH 5.5 μL,37 ℃温育15 min,加入10 mol/L乙酸铵33 μL和冰无水乙醇270 μL于-20 ℃沉淀过夜,然后4 ℃,12 000 r/min离心弃上清液(半径6.5 cm),用70%乙醇洗涤后将沉淀溶于20 μL去离子水中,于-20 ℃冰箱中保存待用。
1.2.4BRCA1的甲基化特异性聚合酶链反应(methylation specific PCR, MSP)
用亚硫酸氢盐修饰 的DNA作模板进行PCR扩增,BRCA1引物参照文献[8]设计(表1)。PCR反应条件: 95 ℃预变性10 min;94 ℃变性1 min,60 ℃退火30 s,72 ℃延伸30 s,35个循环;72 ℃终延伸10 min。扩增结果用含溴化乙锭的2%琼脂糖凝胶电泳检测。用凝胶成像自动分析系统观察结果。判定标准:观察纯化后的DNA以不同引物扩增的产物,若用甲基化引物可扩出产物,而未甲基化引物不能扩出者,计为“+”,即完全甲基化;反之计为“-”,为完全未甲基化;均有产物者计为“±”,即部分甲基化。
1.2.5MS基因的检测
方法同1.2.2,MS基因反应条件:94 ℃预变性5 min;94 ℃变性30 s,57 ℃退火30 s,72 ℃延伸30 s,35个循环;72 ℃终延伸7 min。
1.3 统计学处理
采用GraphPad Prism 5和SPSS 11.0统计软件进行统计学数据分析,数据用均数±标准差()表示。计数资料采用χ2检验,两个独立样本采用t检验,MS基因表达与BRCA1甲基化的相关性用Spearman相关分析,P<0.05为差异有统计学意义。
2 结 果
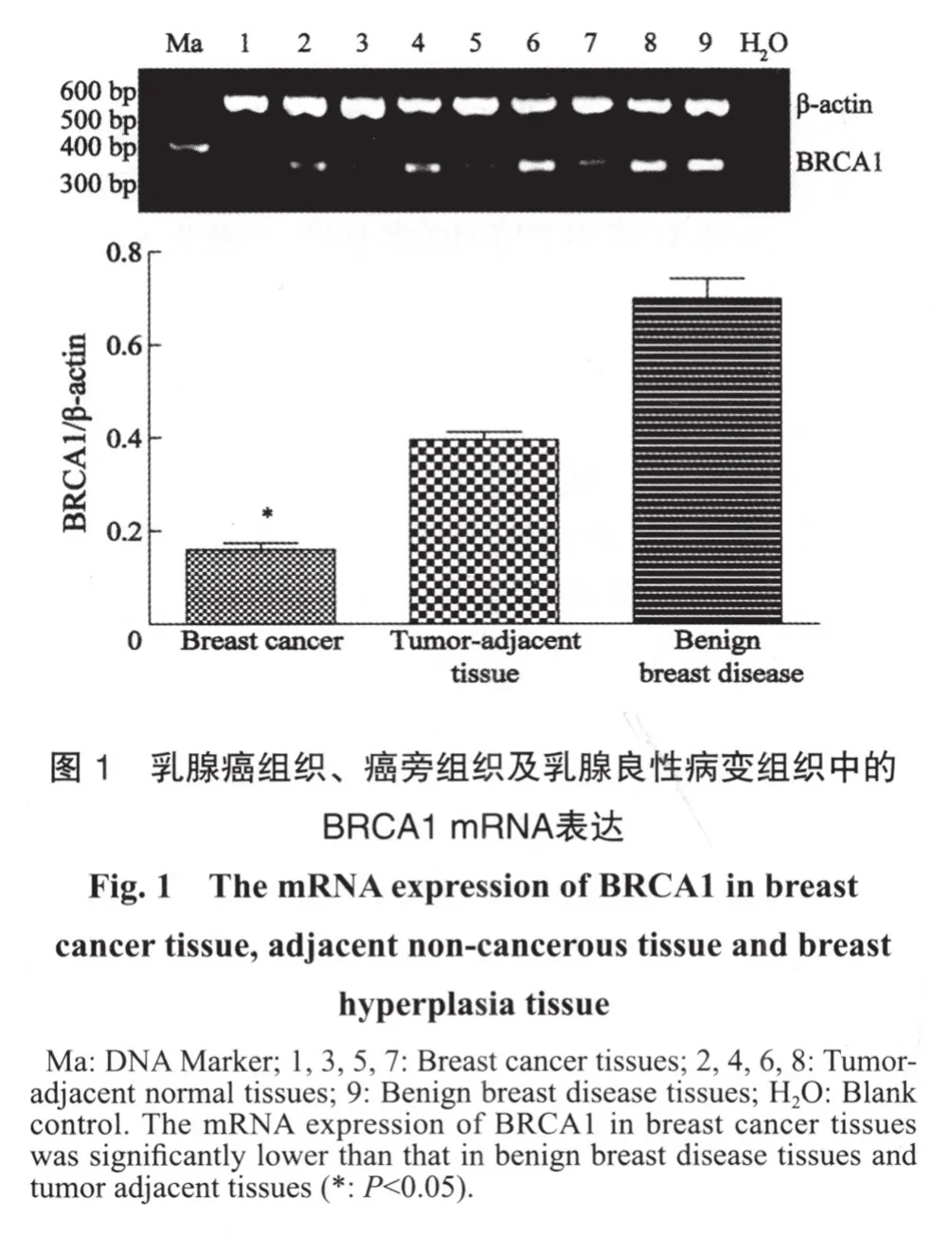
2.1 组织中BRCA1 mRNA表达
乳腺良性病变组织BRCA1 mRNA均有表达,乳腺癌组织、癌旁组织BRCA1 mRNA出现低表达或缺失,31例乳腺癌组织中,17例BRCA1 mRNA表达下降或缺失。其mRNA的表达量用组织中的mRNA的D值与内参D值的比值表示,结果显示乳腺癌组织BRCA1 mRNA表达量(0.16±0.08)明显低于乳腺癌旁组织表达量(0.41±0.09)及乳腺良性病变组织表达量(0.69±0.14),差异均有统计学意义(P<0.05,图1)。
2.2 BRCA1基因启动子甲基化状态
乳腺良性病变组织中未发现BRCA1甲基化,BRCA1基因在31例乳腺癌及癌旁组织中甲基化检出率分别为29.0%(9/31)和3.2%(1/31),经统计学分析,差异有统计学意义(χ2=7.631,P<0.05,图2)。BRCA1甲基化与散发性乳腺癌组织学分级、E R阴性有关(P<0.05),但与发病年龄、组织分型和淋巴结转移无关(P>0.05,表2)。
2.3 BRCA1 mRNA表达与其甲基化的关系
在18例BRCA1 mRNA表达阴性的乳腺癌组织中,有8例BRCA1基因发生了甲基化,在13例BRCA1 mRNA表达阳性的乳腺癌组织中有1例BRC A1基因发生甲基化,两者相比,差异有统计学意义(χ2=4.949,P<0.05)。

表 1 所用引物序列Tab.1 Primer sequences and PCR conditions

2.4 MS mRNA的表达与乳腺癌的关系
乳腺良性病变组织中MS mRNA有表 达,乳腺癌组织及癌旁组织中MS mRNA低表达或缺失(图3),其mRNA的表达量用 组 织中的mRNA的D值与内参D值的比值表示。结果显示,乳腺癌组织的表达量(0.26±0.06)明显低于乳腺良性病变组织的MS mRNA表达量(0.60±0.21)及癌旁组织的表达量(0.40±0.13),差异均有统计学意义(P<0.05,图3)。

表 2 乳腺癌组织BRCA1甲基化与临床资料的关系Tab.2 Relationship between BRCA1 methylation in breast cancer tissues and clinical features

2.5 MS基因表达与BRCA1基因甲基化的关系
MS基因表达阴性的14例乳腺癌组织中,有7例BRCA1基因发生了甲基化;MS基因表达阳性的17例乳腺癌组织中,有2例其BRCA1发生甲基化,经Spearman相关分析结果显示,乳腺癌组织中MS mRNA的表达与BRCA1基因甲基化有相关性(r=0.419,P<0.05)。
3 讨 论
乳腺癌是女性最常见的恶性肿瘤之一,严重威胁着女性的健康。BRCA1基因主要功能是在细胞增殖周期的整个环节对细胞分裂和生长进行调控,使细胞按照正常程序周期性分 裂、生长和凋亡[9-10],是重要的乳腺癌抑癌基因。抑癌基因失活可以导致细胞周期失控,细胞发生异常增殖而癌变。本实验检测到BRCA1基因在乳腺癌组织中表达明显低于癌旁组织及乳腺良性病变组织,提示该基因的失表达与乳腺癌患病密切相关。大量研究已证实甲基化是除基因突变和缺失以外导致抑癌基因失活的第三种机制,在肿瘤的发生、发展中起重要的作用。研究已发现许多抑癌基因通过DNA异常甲基化引起表达抑制甚至关闭而失活,如Rb、APC、MGMT、P16等。我们检测到BRCA1基因乳腺癌组织甲基化检出率为29%,这与之前报道的用不同方法检测到9%~44%的乳腺癌样本中BRCA1基因启动子甲基化结果较一致,癌组织甲基化检出率显著高于相应癌旁组织(3.2%)及乳腺良性病变组织(0)[11-12]。曾有报道提出,BRCA1基因甲基化的肿瘤可能表型模拟家族BRCA1基因肿瘤[13]。我们观察到BRCA1甲基化与散发性乳腺癌组织学分级、ER负性有关,这与Birgisdottir等[5]报道的结果相一致。然而Matros等[6]报道BRCA1基因在高级别ER阳性肿瘤的启动子甲基化频率高,这意味着其表型的关联可能更加复杂。经统计,BRCA1基因的表达与该基因发生甲基化密切相关,DNA甲基化机制可以较合理地解释散发性乳腺癌的BRCA1基因转录水平异常。
癌症基因组学的显著特点是全基因组的低甲基化,和特定位点的高甲基化并存。DNA低甲基化可以提高特定原癌基因(如与增殖和转移相关基因)的表达,而特定位点高甲基化可以使抑癌基因(如DNA修复基因)的表达降低,从而增加患癌症的风险[14],而启动这些变化的机制尚待研究。基因启动子区甲基化模式的改变,是在转录水平调节基因表达的途径之一[15]。大量研究表明,正常细胞中抑癌基因启动子是低甲基化甚至非甲基化的。然而,肿瘤细胞中抑癌基因却常发生甲基化,从而导致抑癌基因低表达和癌细胞增殖能力提高[16-18]。MS主要生化功能是催化同型半胱氨酸复甲基为甲硫氨酸,甲硫氨酸在转甲基之前,与ATP作用生成SAM,为DNA的甲基化提供甲基,是参与甲基供体生成的关键酶。大量实验已证明MS多态性与乳腺癌患病有关,本实验室也发现MS2756等 位基因G型突变能够增加乳腺癌的患病风险,并推测这可能与MS多态性的改变影响了酶的活性,从而影响了甲基供体生成,导致甲基化异常有关[19]。而本 实验进一步检测到乳腺良性病变组织中MS mRNA均有表达,乳腺癌患者的癌组织及癌旁组织中MS低表达或缺失,提示MS基因低表达能增加乳腺癌患病风险 。本实验结果显示,乳腺癌组织中MS mRNA的表达与BRCA1基因甲基化有相关性,这表明MS基因的表达可能影响某些基因的甲基化状态。我们推测MS基因低表达,会影响体内甲基供体生成,导致甲基化异常,从而引起BRCA1基因低甲基化。但我们却发现MS基因表达阴性癌组织中BRCA1基因发生甲基化的频率较表达阳性的显著升高,表明MS基因低表达与抑癌基因BRCA1甲基化有关,符合肿瘤细胞抑癌基因常常发生甲基化这一表观遗传学特征。Stempak等[20]、Kim 等[21]、Gaia等[22]研究表明,甲基供体缺乏对甲基化的影响是位点和基因特异性的,此外,甲基化的变化方向可能是细胞、靶器官、转化具体阶段特异性的,而且未必 是基因组和基因之间或位点的DNA甲基化相同。具体机制还有待于进一步研究。近年来,DNA甲基化模式异常已被确立为人类癌症的标志之一。甲硫氨酸循环关键酶低表达,导致体内甲基化水 平异常,抑癌基因CpG岛高甲基化而失活,从而引发癌症,我们得到的结果初步证实了这一假设。我们的研究结果强调了甲硫氨酸循环对乳腺癌发病的重要性。虽然该循环酶网络十分复杂,但评估限速酶及其与环境的相互作用对于评估乳腺癌风险是十分关键的。
[1]Ehrlich M.DNA methylation in cancer: Too much, but also too little [J].Oncogene, 2002, 21(35): 5400-5413.
[2]Lu M, Wang F, Qiu J.Methionine synthase A2756G polymorphism and breast cancer risk: a meta-analysis involving 18, 953 subjects [J].Breast Cancer Res Treat,2010, 123: 213-217.
[3]Xinran X, Jia C.One-carbon metabolism and breast cancer:an epidemiological perspective [J].J Genet Genomics,2009, 36: 203-214.
[4]Niwa Y, Ciyama T, Nakajima T.BRCAl expression status in relation to DNA methylation of BRCA1 promoter region in sporadic breast cancers [J].Jpn J Cancer Res, 2000, 91(5):519-526.
[5]Birgisdottir V, Stefansson OA, Bodvarsdottir SK, et al.Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer [J].Breast Cancer Res, 2006, 8:R38.
[6]Matros E, Wang ZC, Lodeiro G, et al.BRCA1 promoter methylation in sporadic breast tumors: relationship to gene expression profiles [J].Breast Cancer Res Treat, 2005, 91:179-186.
[7]Esteller M, Corn PG, Baylin SB, et al.A gene hypermethylation profile of human cancer [J].Cancer Res, 2001, 61(8):3225-3229.
[8]Esteller M, Silva JM, Dominguez G, et al.Promoter hypermethylation and BRCA1 inactivation in sporadic breast cancer and ovarian tumors [J].J Natl Cancer Inst(Bethesda), 2000, 92(7): 564-569.
[9]Venkitaraman AR.Cancer susceptibility and the functions of BRCA1 and BRCA2 [J].Cell, 2002, 108: 171-182.
[10]Paull TT, Cortez D, Bowers B, et al.Direct DNA binding by Brca1 [J].Proc Natl Acad Sci U S A, 2001, 98: 6086-6091.
[11]Birgisdottir V, Stefansson OA, Bodvarsdottir SK, et al.Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer [J].Breast Cancer Res, 2006, 8(4):R38.
[12]Butcher DT, Rodenhiser DI.Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours[J].Eur J Cancer, 2007, 43(1): 210-219.
[13]Turner N, Tutt A, Ashworth A.Hallmarks of 'BRCAness' in sporadic cancers [J].Nat Rev Cancer, 2004, 4: 814-819.
[14]Duthie SJ.Epigenetic modifications and human pathologies:cancer and CVD [J].Proc Nutr Soc, 2011, 70(1): 47-56.
[15]Turner JD, Pelascini LP, Macedo JA.et al.Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms [J].Nucleic Acids Res, 2008, 36: 7207-7218.
[16]Taghavi N, Biramijamal F, Sotoudeh M.et al.p16INK4a hypermethylation and p53, p16 and MDM2 protein expression in esophageal squamous cell carcinoma [J].BMC Cancer,2010, 10: 138.
[17]Gonzalo V, Lozano JJ, Muñoz J.et al.Aberrant gene promoter methylation associated with sporadic multiple colorectal cancer [J].PLoS One, 2010, 5: e8777.
[18]Wang X, Lau KK, So LK, et al.CHD5 is down-regulated through promoter hypermethylation in gastric cancer [J].J Biomed Sci, 2009, 16: 95.
[19]韩琳琳, 侯琳, 宋金莲.甲硫氨酸合成酶多态性及CHD5基因甲基化与乳腺癌发病的关系[J].中国癌症杂志,2010, 20(4): 251-255.
[20]Stempak JM, Sohn KJ, Chiang EP, et al.Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model [J].Carcinogenesis, 2005, 26: 981-990.
[21]Kim YI.Nutritional epigenetics: impact of folate deficiency on DNA methylation and colon cancer susceptibility [J].J Nutr, 2005, 135(11): 2703-2709.
[22]Gaia B, Erika V, Sei-Ichi M, et al.Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells [J].BMC Biol, 2010, 8: 6.
猜你喜欢
中国生物化学与分子生物学报(2022年7期)2022-09-07
岭南现代临床外科(2022年1期)2022-03-16
中国药科大学学报(2021年6期)2021-12-31
中国男科学杂志(2016年9期)2016-03-20
中国医疗美容(2015年1期)2015-07-12
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
