
南五味子、五味子HPLC指纹图谱研究和木脂素成分测定
2011-05-26 07:17李晓亮易进海刘云华朱美晓
中成药 2011年6期
李晓亮, 易进海, 刘云华, 朱美晓, 吴 燕
(1.泸州医学院,四川泸州 646000;2.四川省中医药科学院,四川成都 610041;3.成都中医药大学,四川成都 611137)
南五味子为木兰科植物华中五味子Schisandra sphenantheraRehd.et Wils.的干燥成熟果实,五味子为木兰科植物五味子Schisandra chinensis(Turcz)Baill.的干燥成熟果实,二者具有相同的功能与主治[1]。现行版中国药典分别以五味子酯甲、五味子醇甲作为南五味子、五味子的质量控制指标,这种传统的单一指标的质量控制模式,难以反映药材的真实质量。HPLC指纹图谱技术[2-6]具有适用选择性强、范围广、灵敏度高、分离性能好等优点,是一种评价天然药物一致性和稳定性的综合多要素质量控制方法。谭春梅等[7]、李学龙等[8]对南五味子和五味子指纹图谱进行了研究,但共有峰少、主要色谱峰分离效果差。本实验经过大量优化实验所建立的HPLC指纹图谱,分析时间短,共有峰多且主要定量峰达到基线分离,兼具有定性和定量的作用,为全面控制南五味子、五味子药材质量提供科学方法。
1 仪器与试药
Agilent 1200型高效液相色谱仪(包括四元泵,DAD检测器,柱温箱,自动进样器,工作站);KQ-100超声波清洗仪(昆山市超声仪器有限公司);岛津AUW220D型十万分之一电子天平。乙腈、四氢呋喃为美国Tedia色谱纯;水为乐百氏纯净水;其余试剂均为分析纯。
五味子酯甲对照品(批号:1529-200001)、五味子甲素对照品(批号:0764-200107)、五味子醇甲对照品(批号:1529-200001)、五味子乙素对照品(批号:110765-200609)均购自中国药品生物制品检定所。
14批南五味子药材和7批五味子药材经四川省中医药科学院舒光明研究员鉴定分别为木兰科植物华中五味子Schisandra sphenantheraRehd.et Wils.和五味子Schisandra chinensis(Turcz)Baill的干燥成熟果实。
2 方法与结果
2.1 色谱条件 色谱柱:Eclipse XDB-C18分析柱(150 mm ×4.6 mm,5 μm);流动相 A 相为乙腈,B相为水-四氢呋喃(3∶1),梯度洗脱程序:0~35 min,流动相A由8% ~20%,B由92% ~80%;35~45 min,流动相A由20% ~35%,B由80% ~65%;45~55 min,A 为35%,B 为65%,55~65 min,A 由35% ~40%,B由65% ~60%;体积流量1.0 mL/min,柱温35℃,检测波长220 nm。
2.2 对照品溶液的制备 精密称取五味子酯甲对照品 5.33 mg、五味子甲素对照品 5.77 mg、五味子醇甲对照品6.55 mg、五味子乙素对照品4.69 mg,置同一25 mL量瓶中,加甲醇溶解定容至刻度,摇匀,制成五味子酯甲、五味子甲素、五味子醇甲、五味子乙素的混合对照品溶液,质量浓度分别为213.2 mg/L、230.8 mg/L、262.0 mg/L、187.6 mg/L。
2.3 供试品溶液的制备 精密称取南五味子、五味子药材粉末(过三号筛)约1.0 g,置50 mL具塞锥形瓶中,精密加甲醇25 mL,称定质量,超声处理30 min(100 W,40 kHz),放冷,再称定质量,用甲醇补足减失质量,摇匀,滤过,取续滤液,即得。
3 南五味子、五味子指纹图谱研究
3.1 精密度实验 精密吸取同一供试品溶液(南五味子S1),重复进样5次,结果各共有峰的相对保留时间RSD<0.4%,相对峰面积RSD<1.5%,相似度为0.99,表明仪器精密度好。
3.2 稳定性实验 精密吸取同一供试品溶液(南五味子 S1),分别于0、2、4、8、16、24 h 测定,结果各共有峰的相对保留时间RSD<0.4%,相对峰面积RSD<1.4%,相似度为0.98,表明供试品溶液于24 h内测定稳定。
3.3 重复性实验 取同批样品(南五味子S1)5份,精密称定,分别制备供试品溶液进行分析,结果各共有峰的相对保留时间RSD<0.6%,相对峰面积RSD<1.6%,相似度为0.99,表明该实验方法重复性好。
3.4 南五味子、五味子指纹图谱的建立
3.4.1 南五味子药材色谱指纹图谱共有模式的建立 取不同来源的南五味子药材(S1~S14)粉末,精密称定,按2.3项下方法制备供试品溶液,精密吸取10 μL,分别注入液相色谱仪,按2.1项下色谱条件测定,记录65 min的HPLC图,采用国家药典委员会“中药色谱指纹图谱相似度评价系统(2004A)”进行数据分析处理,设定S1为参照图谱,将其它样品的色谱峰与参照图谱进行自动匹配,生成南五味子药材指纹图谱共有模式(即对照图谱,见图1),计算1~14批样品与对照图谱相似度,分别为0.986、0.994、0.988、0.988、0.996、0.995、0.990、0.965、0.981、0.985、0.932、0.932、0.977、0.977。14 批南五味子药材指纹图谱与对照图谱之间相似度均大于0.9,说明相似度良好。见图2。

图1 南五味子对照图谱Fig.1 The reference map of Schisandrae sphenantherae Fructus
3.4.2 五味子药材色谱指纹图谱共有模式的建立
同3.4.1项下方法建立五味子药材指纹图谱共有模式(即对照图谱,见图3),计算1~7批样品与对照图谱相似度,分别为 0.998、0.998、0.999、0.999、0.999、0.997、0.997。7批五味子药材指纹图谱与对照图谱之间相似度均大于0.9,说明相似度良好。见图4。

图2 南五味子指纹图谱Fig.2 Fingerprints of Schisandrae sphenantherae Fructus
3.4.3 共有指纹峰的标定 将南五味子及五味子药材的指纹图谱峰锁定在5 min~65 min共有指纹区进行统计,采用相对保留时间从14批南五味子样品图谱的共有指纹区标定了25个共有峰,从7批五味子样品图谱的共有指纹区标定了24个共有峰,分别经过与对照品比对并结合DAD光谱,可知1号峰为五味子醇甲,12号峰为五味子酯甲,15号峰为五味子甲素,21号峰为五味子乙素(图1、图3)。14批南五味子样品及7批五味子样品出峰总数及共有峰占总峰面积百分比见表1。

图3 五味子对照图谱Fig.3 The reference map of Schisandrae chinensis Fructus

表1 样品来源及指纹图谱数据Tab.1 Sources of samples and data of HPLC fingerprint
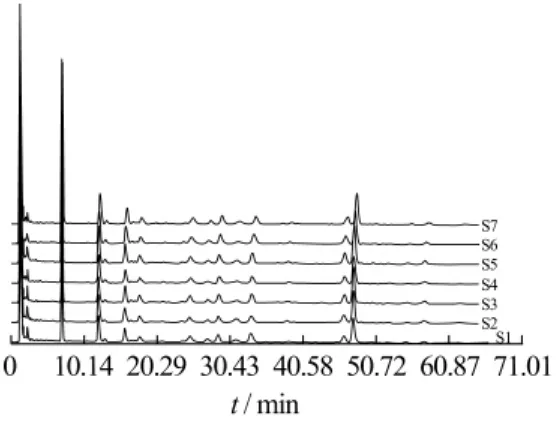
图4 五味子指纹图谱Fig.4 Fingerprints of Schisandrae chinensis Fructus
3.4.4 南五味子、五味子指纹图谱比较 采用国家药典委员会“中药色谱指纹图谱相似度评价系统(2004B)”进行数据分析处理,以南五味子对照图谱作为参照图谱,计算14批南五味子和7批北五味子图谱的相似度,结果见表1,表明南五味子、五味子指纹图谱差异很大,显著不同。南五味子中五味子酯甲、五味子甲素的量远大于五味子,而五味子中五味子醇甲的量远大于南五味子,但二者总木脂素量相当,具体数据见表2。
4 南五味子、五味子中4种木脂素的测定
4.1 系统适用性实验 按2.1项下色谱条件进行检测,五味子醇甲、五味子酯甲、五味子甲素及五味子乙素与相邻色谱峰分离度均大于1.5。理论塔板数按五味子酯甲计大于5 000。对照品溶液及供试品溶液的色谱图见图5。

表2 五味子酯甲、五味子甲素、五味子醇甲、五味子乙素的质量分数Tab.2 Contents of deoxyschizandrin,schisantherrin A,Schisandrin and γ-schisandrin

图5 对照品及药材样品HPLC色谱图Fig.5 HPLC chromatograms of control solution and sample solution
4.2 线性关系 精密吸取对照品溶液2.0、4.0、6.0、8.0、10.0、12.0 μL 依次分别注入液相色谱仪,测定峰面积,以对照品进样量(μg)为横坐标X,峰面积(A)为纵坐标Y,绘制标准曲线,得回归方程,五味子酯甲:Y=5 164.06X+1.73,r=0.999 9;五味子甲素:Y=6 006.40X+1.65,r=0.999 9;五味子醇甲:Y=5 832.03X+1.75,r=0.999 9;五味子乙素Y=5 316.11X+1.69,r=0.999 9。结果表明五味子酯甲、五味子甲素、五味子醇甲、五味子乙素分别在 0.426 4 ~ 2.558 μg、0.461 6 ~ 2.770 μg、0.524 0 ~3.144 μg、0.375 2 ~2.251 μg 范围呈良好的线性关系。
4.3 精密度实验 精密量取混合对照品溶液10 μL,连续进样5次,按2.1项下色谱条件进行检测,记录峰面积。五味子酯甲、五味子甲素、五味子醇甲、五味子醇乙峰面积RSD分别为0.3%、0.2%、0.6%、0.3%,表明仪器精密度良好。
4.4 重复性实验 取同批次药材粉末5份,照2.2项下方法制供试品溶液,依法测定峰面积,五味子酯甲、五味子甲素、五味子醇甲、五味子乙素含量RSD分别为0.3%、0.9%、0.4%、0.5%,表明该方法的重复性良好。
4.5 稳定性实验 取供试品溶液于室温放置,分别于0、1、2、4、8、16、24 h 依法测定,记录峰面积。五味子酯甲、五味子甲素、五味子醇甲、五味子乙素峰面积 RSD 分别为 0.3%、0.2%、0.6%、0.3%,表明供试品溶液稳定性良好。
4.6 回收率实验 精密称取上述已知质量分数同批次药材粉末0.1 g,5份,分别精密加入五味子酯甲(213.2 mg/L)、五味子甲素(230.8 mg/L)、五味子醇甲(262.0 mg/L)、五味子乙素(187.6 mg/L)对照品溶液各2.5 mL,按2.2项下方法制成供试品溶液。依法测定,计算回收率,五味子酯甲、五味子甲素、五味子醇甲、五味子乙素平均回收率分别为97.5%(RSD 为 2.3%)、94.8%(RSD 为 3.1%)、103.7%(RSD 为 1.6%)、98.2%(RSD 为 1.1%)。4.7 测定 取各批次药材粉末(过三号筛)约0.2 g,分别依2.3项下方法制备成供试品溶液,依法测定,结果见表2。
5 讨论
5.1 本实验比较了文献报道的多种流动相系统,甲醇-水系统、乙腈-水系统等[8-12]分离效果均不理想。本实验在乙腈-水系统中加入适量的四氢呋喃,比较了Eclipse XDB-C18色谱柱(150 mm × 4.6 mm,5 μm)和 Synergi Hydro-rp C18色谱柱(150 mm × 4.6 mm,4 μm),在该色谱条件下梯度洗脱时间大大缩短,分离得到25个共有峰且主要定量峰达到基线分离,兼具有定性和定量的作用,系统适用性良好。
5.2 本实验研究结果显示不同产地南五味子指纹图谱相似度大于0.9,但其主要木脂素成分量差异较大;不同产地五味子指纹图谱相似度亦大于0.9,但南五味子、五味子指纹图谱相似度小于0.3,表明南五味子、五味子指纹图谱特征性和专属性强。南五味子中五味子酯甲和五味子甲素质量分数远大于五味子,而五味子中五味子醇甲质量分数远大于南五味子,但二者木脂素总量相当。因此,指纹图谱结合木脂素成分的测定,可为全面控制南五味子、五味子药材的质量提供科学方法。
[1]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:227.
[2]谢 青.现代色谱技术在中药指纹图谱研究中的应用[J].海峡药学,2007,19(11):95-98.
[3]胡育筑,孟庆华,刘永锁,等.中药色谱指纹谱的质量控制方法[J].色谱,2004,2(24):361.
[4]谢培山.中药色谱指纹图谱[M].北京:人民卫生出版社,2005:124.
[5]王丽聪,曹玉华,徐红兰,等.银黄口服液的质量控制及其高效液相色谱指纹图谱的研究[J].色谱,2006,24(4):367.
[6]马丽萍,丘小惠,闵 江,等.高良姜饮片HPLC指纹图谱及高良姜素含量测定研究[J].中成药,2008,30(11):1565-1569.
[7]谭春梅,黄琴伟,张文婷,等.五味子和南五味子的HPLC指纹图谱研究[J].中国现代应用药学杂志,2008,25(6):514-517.
[8]李学龙,杨 博,徐 鹏,等.南五味子液相色谱指纹图谱研究[J].大连工业大学学报,2009,28(1):4-8.
[9]代云桃,金高娃,秦雪梅,等.五味子提取物高效液相色谱分析方法的优化[J].色谱,2009,27(4):442-446.
[10]宋平顺,丁永辉,赵建邦.南五味子不同炮制品中五味子酯甲和五味子甲素含量的 HPLC法测定[J].中药材,2008,31(5):652-654.
[11]耿立超,杨中林.HPLC法测定不同产地五味子中木脂素的含量[J].中医药学报,2007,35(2):47-48.
[12]杨 博,陈 彤,李学龙,等.高效液相色谱法测定南、北五味子中木脂素含量[J].大连工业大学学报,2009,28(2):102-106.
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2021年10期)2021-12-02
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年4期)2018-05-07
中成药(2017年9期)2017-12-19
中成药(2017年3期)2017-05-17
中成药(2017年3期)2017-05-17
中国继续医学教育(2015年2期)2016-01-06
药学研究(2015年11期)2015-12-19
中国药理学通报(2014年2期)2014-05-09
