
百蕊草药材的指纹图谱研究
2011-05-26 07:57许伏新徐国兵韩玲玲方成武刘守金梁益敏
中成药 2011年7期
汤 超, 许伏新, 徐国兵, 韩玲玲, 方成武, 刘守金, 梁益敏
(1.安徽医科大学药学院,安徽合肥230032;2.安徽省食品药品监督管理局,安徽 合肥230023;3.安徽省食品药品检验所,安徽合肥230051;4.安徽中医学院,安徽合肥 230038)
百蕊草药材的指纹图谱研究
汤 超1, 许伏新2*, 徐国兵3, 韩玲玲3, 方成武4, 刘守金4, 梁益敏4
(1.安徽医科大学药学院,安徽合肥230032;2.安徽省食品药品监督管理局,安徽 合肥230023;3.安徽省食品药品检验所,安徽合肥230051;4.安徽中医学院,安徽合肥 230038)
目的 采用高效液相色谱法建立来自14个不同产地的15批百蕊草样品的指纹图谱。方法 甲醇加热回流提取1 h获得百蕊草提取液,采用HPLC分析百蕊草的指纹图谱。色谱柱Inertsil OSD-SP(4.6 mm×150 mm,5 μm);流动相甲醇-0.4%磷酸水溶液梯度洗脱;分析时间75 min;检测波长365 nm;柱温30℃;体积流量0.8 mL/min。结果 建立了百蕊草的指纹图谱,5号山柰素峰为对照峰,确定了8个共有峰。结论 该指纹图谱方法简单,重现性好,可作为百蕊草药材质量控制的重要方法。
百蕊草;高效液相色谱;指纹图谱
百蕊草Thesium chineseTurcz.为檀香科Santalaceae植物百蕊草的干燥全草,其性味平缓、微苦、涩、寒,具有清热解毒、消肿之功能。临床上多用于治疗急性乳腺炎、肺炎、扁桃体炎、咽喉炎、疖肿等病症[1-2]。现代药理研究表明:百蕊草具有显著的抗炎、解热、镇痛和抗菌作用,其所含的黄酮类化合物为其抗菌的有效成分[3-6]。目前对百蕊草药材的质量控制方法主要是总黄酮的测定[7],以及山柰素的薄层鉴别和测定[8-9]等,为更有效的控制其质量,本实验应用梯度洗脱法建立了百蕊草的HPLC指纹图谱的分析方法,对安徽省各主要百蕊草产地所产药材进行比较性的研究,并以百蕊草药材对照图谱与湖北省采集的百蕊草药材指纹图谱进行了对比研究。
1 仪器与试药
1.1 仪器 岛津LC-20AT高效液相色谱仪(DAD检测器,岛津LC solution色谱工作站);色谱柱为Inertsil OSD-SP(4.6 mm ×150 mm,5 μm);国家药典委员会“中药色谱指纹图谱相似度评价系统”(版本2004);Mettler AE-240型电子天平(万分之一,瑞士Mettler);超声清洗机(美国 Elmasonic)。
1.2 试药 山柰素对照品(批号:110861-200808)购自中国药品生物制品检定所;甲醇(色谱纯,国药集团化学试剂有限公司);水为自制重蒸水;其它试剂均为分析纯。
1.3 百蕊草药材 各百蕊草药材样品具体来源见表1,经安徽中医学院刘守金教授鉴定为檀香科植物百蕊草Thesium chineseTurcz.。

表1 样品来源Tab.1 Sources of sample
2 方法与结果
2.1 供试品溶液的制备 称取0.5 g的百蕊草药材粉末(过80目筛),精密称定,精密加入甲醇25 mL,称定质量,加热回流1 h,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液15 mL,置平底烧瓶中,精密加入50%盐酸5 mL,摇匀,置水浴中加热水解30 min,立即冷却,转移至25 mL量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
2.2 对照品溶液的制备 精密称取山柰素对照0.5 mg,置10 mL量瓶中,加甲醇定容至刻度,摇匀,0.45 μL 微孔滤膜滤过,即得。
2.3 色谱条件 色谱柱:Inertsil OSD-SP(4.6 mm×150 mm,5 μm);流动相:甲醇(A 相)-0.4%磷酸水溶液(B相),梯度洗脱条件:B 0~40 min(80% ~60%);40~70 min(60%);70~75 min(60%~80%);体积流量:0.8 mL/min,柱温:30 ℃,检测波长:365 nm,分析时间:75 min,进样量:20 μL。
2.4 方法学考察
2.4.1 精密度实验 取同一样品,按供试品溶液的制备方法制备,连续进样5次,检测指纹图谱。各共有峰的相对保留时间RSD均小于2%,相对峰面积RSD均小于5%。
2.4.2 重复性实验 取同一批样品5份,按供试品溶液的制备方法制备,连续进样进行分析。各共有峰的相对保留时间RSD均小于2%,相对峰面积RSD均小于5%。
2.4.3 稳定性实验 取同一样品,按供试品溶液的制备方法制备,分别于 0、2、4、8、24 h 进样 5 次,考察色谱峰相对峰面积的一致性。各共有峰的相对保留时间RSD均小于2%,相对峰面积RSD均小于5%,结果表明供试品溶液24 h内稳定性良好。
2.5 样品指纹图谱分析及结果
2.5.1 不同产地百蕊草药材指纹图谱的建立 取各百蕊草药材,按2.1项下方法制备供试品溶液,分别精密吸取20μL注入液相色谱仪,按2.3项下的色谱条件分别进样,记录色谱图,根据测定结果,制定以山柰素为参照物的指纹谱图。对照品图谱见图1,百蕊草不同产地指纹图谱见图2。
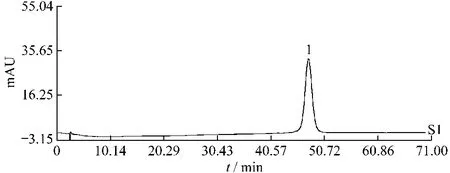
图1 对照品溶液HPLC色谱图Fig.1 HPLC chromatogram of reference substances
2.5.2 百蕊草药材共有指纹峰的建立 通过“中药色谱指纹图谱相似度评价系统”(版本2004)软件对安徽的13批药材的色谱数据进行处理,经过比对,将时间窗宽度设为0.5,选择山柰素的色谱峰进行校正及自动匹配,以中位数法生成百蕊草共有模式指纹图谱(见图3)。13批样品有8个共有峰,其中5号峰为山奈素。以3号色谱峰(保留时间为41.209 min)作为参照峰,计算各共有峰的相对保留时间和相对峰面积,具体数据见表2、表3。各样品出峰总数占总峰面积百分比见表4。
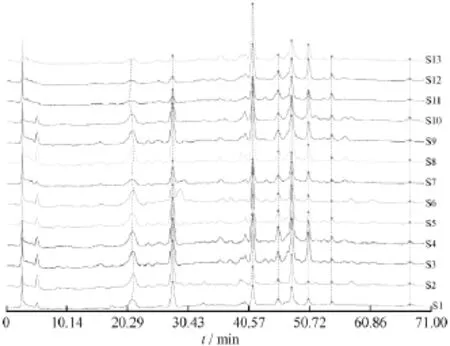
图2 百蕊草HPLC色谱图Fig.2 Fingerprint of Thesinm Chinese Turcz.from Anhui province
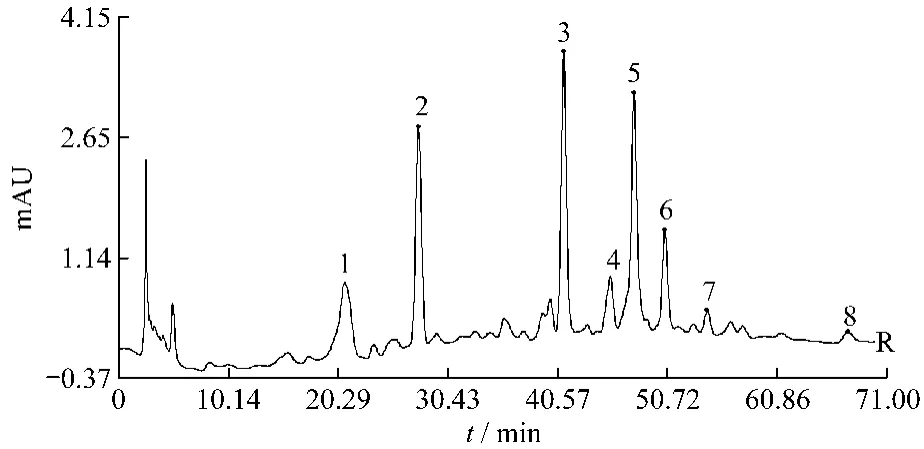
图3 共有模式图Fig.3 Common pattern
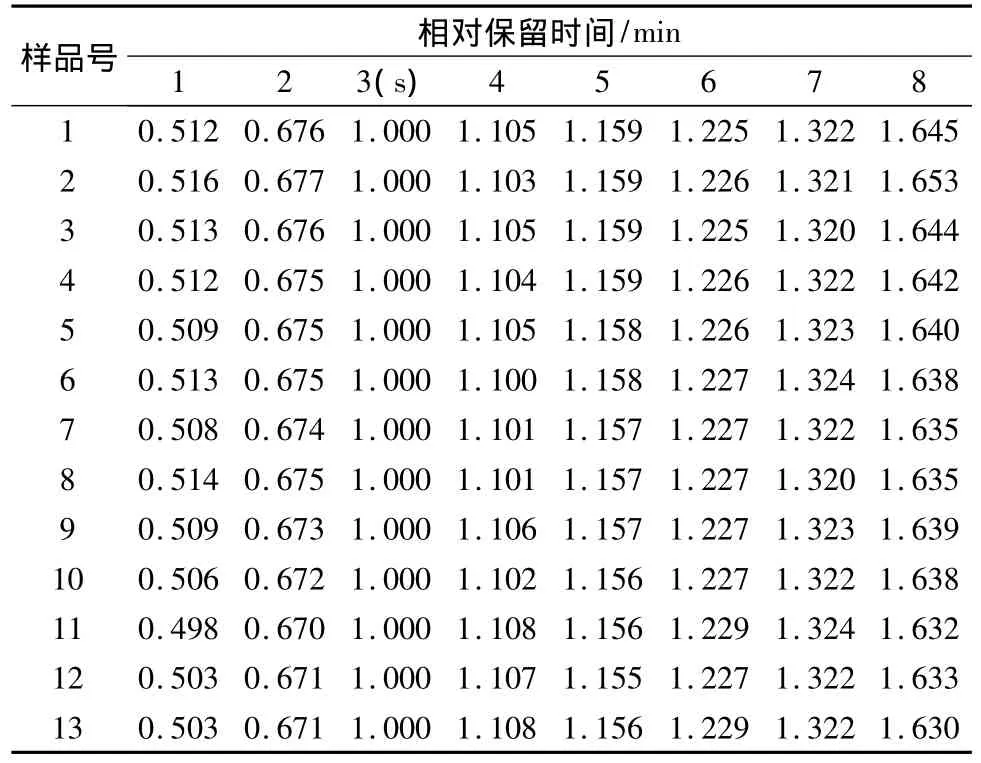
表2 各样品共有峰相对保留时间Tab.2 Relative retention time of common peaks for various samples

表3 各样品共有峰相对峰面积Tab.3 Relative areas of common peaks for various samples
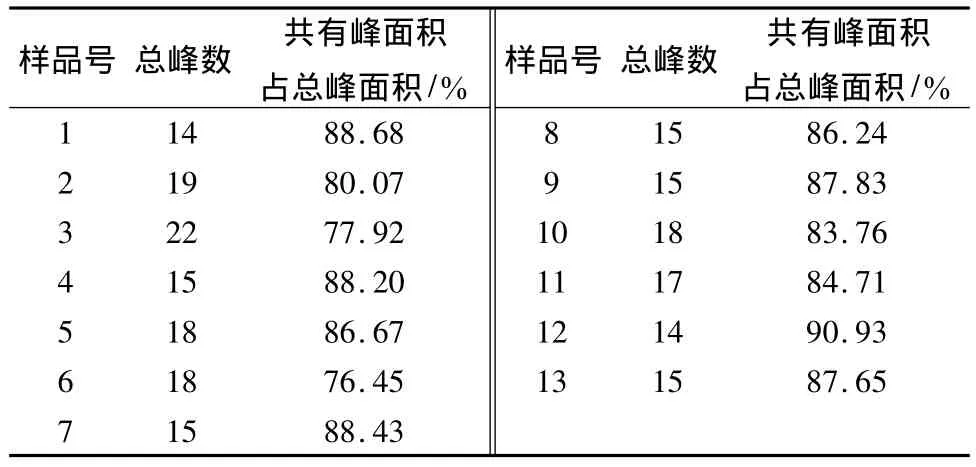
表4 各样品共有峰面积Tab.4 Common peak area for various sample
2.5.3 百蕊草药材相似度评价 通过“中药色谱指纹图谱相似度评价系统”(版本2004)对安徽13批百蕊草药材的色谱数据进行处理,以共有峰为对照模板,计算各色谱图整体的相似度,具体数据见表5。
2.5.4 对其它产地百蕊草药材进行的评价 在相似度计算软件中以百蕊草药材的共有模式图为参照图谱,导入湖北省采集的1年生和2年生药材各1批的指纹图谱,以对照品色谱峰进行校正,自动匹配后计算相似度,结果两批药材相似度分别为0.974、0.914。
3 讨论
3.1 流动相及波长的选择 实验研究过程中,通过考察甲醇-水、甲醇-0.4%磷酸水、乙腈-水、乙腈-0.4%磷酸水溶液等多种流动相体系,通过比较,以甲醇-0.4%磷酸水溶液系统洗脱可使色谱峰出峰较好,故选用该流动相系统。本实验前期阶段结合了190~400 nm波段的三维扫描图,对HPLC-DAD色谱图进行分析,选择指标成分山柰素和其它成分紫外吸收相对均较强的波长,结果在365 nm处山柰素的吸收较强,且色谱图各色谱峰信号较丰富,所以选定365 nm为检测波长。

表5 样品相似度Tab.5 Similarity of samples
3.2 样品提取条件的筛选 对百蕊草药材提取方法进行研究时,试验了不同提取溶剂[10](95%乙醇、50%乙醇、30%乙醇、50%甲醇、甲醇),以及超声提取、回流提取、索氏提取等不同提取方式,同时还考察了不同提取时间的提取效果[11-12]。通过一系列的实验表明,用甲醇回流提取1 h提取液的成分最多,信息量最丰富,因此选用甲醇回流提取1 h的方法进行提取。
3.3 百蕊草指纹图谱的相似度分析 采用了HPLC法对百蕊草药材甲醇提取物进行分析,各样品HPLC指纹图谱虽存在一定差异,但均具有相同的色谱特征峰;除安徽滁州三批药材相似度评价结果小于0.9外,安徽其它产地10批样品相似度均大于0.9,说明本研究所得的百蕊草色谱指纹图谱共有模式具有较强的专属性。
应用“中药色谱指纹图谱相似度评价系统”(版本2004)对滁州三批进行相似度评价,相似度分别为0.959,0.990,0.972,说明滁州产百蕊草药材与安徽其它产地的百蕊草药材质量存在一定的差异。
在指纹图谱相似度大于0.9的药材色谱图上,各峰的归一化峰面积结果表明,不同产地百蕊草药材主要成分的质量分数仍然存在明显差异,建议在色谱指纹图谱相似度评价的基础上,结合质量标准中的鉴别、检查、浸出物、主成分定量测定等项目,进行百蕊草药材的综合评价,以保证临床用药的安全有效。
[1]卫生部药典委员会.中国药典:一部[S].北京:人民卫生出版社,1977:215-216.
[2]国家中医药管理局《中华本草》编委会.中华本草[M].上海:上海科学术出版社,1999:595-597.
[3]曹明成.百蕊草提取工艺研究[J].现代中药研究与实践,2004,18(1):57-58.
[4]刘永松,潘 玲,祁克宗,等.百蕊草有效提取成分对七种细菌的敏感性试验[J].贵州医药,2006,30(6):564-566.
[5]丁秀年,张三军,明 亮.百蕊草含片对小鼠的镇痛作用[J].淮海医药,2001,19(1):17-18.
[6]杨 军,王 静,高美华.百蕊草片药理作用的实验研究[J].中国中药杂志,1999,24(6):367-369.
[7]徐雷鸣,汪素兰.紫外分光光度法测定百蕊草总黄酮含量[J].基层中药杂志,1995,9(4):29-31.
[8]徐国兵,黄万著,王德群,等.百蕊草药材中山奈酚和总黄酮的含量测定[J].中医药学报,2008,36(1):39-41.
[9]刘本俊,武承英.百蕊草口服液制备工艺比较研究[J].时珍国医国药,2000,11(8):688-689.
[10]钟方丽,王晓林,纪萍萍,等.百蕊草中总黄酮的提取工艺研究[J].吉林化工学院学报,2008,25(4):5-7.
[11]冯年平,范广平,吴春兰,等.微波萃取技术在中药提取中的应用[J].世界科学技术:中药现代化,2002,4(2):49-52.
[12]王 平,刘川生,章银军,等.微波辅助萃取姜黄素的研究[J].中国天然药物,2004,2(5):319-320.
HPLC fingerprint of Thesium chinese Turcz.
TANG Chao1, XU Fu-xin2*, XU Guo-bing3, HAN Ling-ling3, FANG Cheng-wu4, LIU Shou-jin4,LIANG Yi-min4
(1.College of Pharmaceutical Sciences,Anhui Medical University,Hefei 230032,China;2.Anhui Provincial Food and Drug Administration,Hefei 230023,China;3.Anhui Institute for Food and Drug Control,Hefei 230051,China;4.Anhui College of Traditional Chinese Medicine,Hefei 230038,China)
AIMTo establish chromatographic fingerprints of fifteen batches ofThesium chineseTurcz.from fourteen different habitats by HPLC.METHODSThe extract ofThesium chineseTurcz.was obtained with methyl hydrate by thermal reflux for an hour.Analysis was performed on Inertsil OSD-SP column(4.6 mm ×150 mm,5 μm)with gradient mobile phase of methanol-0.4%H3PO4.The fingerprint finished in 75 min,the detection wavelength was at 365 nm and the column temperature was 30 ℃ with the flow rate of 0.8 mL/min.RESULTSEstablishing the fingerprints ofThesium chineseTurcz.,marker peak was kaempferide(No.5 peak),eight common peaks were found in the HPLC fingerprints from different habitats.CONCLUSIONThe method is simple,accurate,with good reproducibility,and it can be used specifically for the quality control ofThesium ChineseTurcz.
Thesium chineseTurcz.;fingerprint;HPLC
R284.1
A
1001-1528(2011)07-1102-04
2010-08-31
国家中医药管理局中医药科学技术研究专项(06-07ZP25);安徽省学术和技术带头人及后备人选科研活动经费资助项目(2009年)
汤 超(1986—),男,硕士生。Tel:13966766557,E-mail:tiankong611@yahoo.com.cn
* 通信作者:许伏新(1968—),男,硕士,从事药事管理研究。Tel:(0551)2999221,E-mail:fuxin@ada.gov.cn
猜你喜欢
今日农业(2022年2期)2022-11-16
今日农业(2021年6期)2021-06-09
特种经济动植物(2021年4期)2021-04-19
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
中国卫生(2016年6期)2016-11-23
中国卫生(2016年8期)2016-11-12
中国卫生(2016年8期)2016-11-12
中国卫生(2016年5期)2016-11-12
自动化学报(2016年8期)2016-04-16
