
中间相炭微球为炭源反应形成SiC陶瓷及其结构与性能①
2011-05-03 08:29:32夏鸿雁王继平乔冠军
固体火箭技术 2011年4期
夏鸿雁,王继平,黄 斌,乔冠军
(西安交通大学金属材料强度国家重点实验室,西安 710049)
0 引言
SiC陶瓷具有优异的热物理性能、高机械强度、耐腐蚀性及抗氧化性等,是一种具有广泛应用前景的工程陶瓷材料。反应形成法具有烧结温度低、周期短的特点及制备异形件的能力,成为制备SiC陶瓷方法中很有吸引力的一种。该方法涉及多孔炭预制体的制备、液相硅的渗入及Si/C反应生成SiC。目前报道的多孔炭预制体炭源主要是树脂炭、石油焦炭、炭黑等[1-5]。反应形成 SiC陶瓷缺陷在于其制品残硅量高,限制了其在高温及腐蚀环境下的应用。
Calderon等[3]采用不同炭源制备了具有不同活性及孔隙率的预制体,然后高温渗硅制备出反应形成SiC陶瓷。试验结果表明,生成SiC的量明显受到炭材料各向异性程度(即结构有序性)的影响,炭材料的各向异性程度越高,化学活性越强,越易于与硅反应生成SiC。另外,当预制体孔隙率达到36%以上时,液相硅才能够充分渗入,否则会被阻塞而无法充分渗入。中间相炭微球(mesocarbon microbeads,MCMBs)是从中间相沥青基质中分离制备的微米级中间相球体。MCMBs内部呈层状结构,由定向缩聚芳烃堆集而成,即微观结构有序性高[6]。此外,MCMBs具有自粘结性,故在成型时可避免粘结剂的使用,保证了炭源结构的一致性。
本研究以MCMBs为炭源,同时添加SiC粉末抑制MCMBs收缩来调整炭预制体气孔率,最终制备出了反应形成SiC陶瓷块体材料。通过调整烧结温度及保温时间,分析制备的SiC陶瓷各相含量、形貌及性能变化,并进行相应的讨论。
1 试验
首先对MCMBs(平均粒径21μm)进行250℃预氧化处理,然后与SiC粉末(平均粒径40 nm)混合,其中SiC粉末质量含量分别为10%、20%、30%、40%。用玛瑙球将混合物在酒精介质中球磨24 h,烘干,过200目筛。称取一定量的混合物,在室温下100 MPa模压成型,成型尺寸4 mm×5 mm×50 mm。最终以300~400℃/h的升温速率在不同温度(1 500、1 600、1 700℃)和不同保温时间(0、30、60 min)下真空熔融渗硅。根据原料中SiC粉末含量,硅化后制品分别表示为10RFSC、20RFSC、30RFSC和40RFSC。为了观察与了解渗硅前预制体的形貌与孔隙率,本试验另取模压后制品进行1 300℃焙烧处理。
用Archimedes法测定1 300℃煅烧预制体及硅化后SiC陶瓷的密度和开气孔率。采用三点弯曲法测抗弯强度,制品跨距16 mm,加载速度0.5 mm/s。电阻率用四端子法测量。扫描电镜观察硅化前后制品形貌。
2 结果与讨论
2.1 硅化前预制体
预制体开气孔率及形貌对硅化后SiC陶瓷微观结构与性能有重要的影响。图1显示了原料中SiC粉含量对1 300℃煅烧预制体密度及开气孔率的影响。由图1可见,当SiC粉质量含量由10%增至40%时,预制体密度由1.62 g/cm3降至1.32 g/cm3,而开气孔率由22.05%增加到43.78%。MCMBs具有自粘结性,在煅烧过程中会产生均匀收缩,而SiC粉不仅具有烧结惰性,同时随其掺量的增加,制品内部缺陷随之增加,从而抑制制品收缩,起到造孔的作用。
图2是预制体渗硅前微观形貌,图中层片状炭结构清晰可见。根据李同起等[7]报道,热处理温度达到1 000℃以上后获得的MCMB断面呈现出明显的炭层结构,随着热处理温度继续升高,MCMB炭层有逐渐细化和趋于平整的趋势,即结构有序度提高。而这种有序度高的炭结构有利于Si/C反应。另一方面,SiC粉具有催化石墨化的作用,有助于进一步提高炭结构的有序度[8]。

图1 预制体密度、开气孔率随SiC粉含量的变化Fig.1 Variation of density and open porosity of preform s w ith SiC pow der content

图2 预制体表面SEMFig.2 Surface SEM image of preform s
2.2 硅化SiC陶瓷各相含量
在高温熔融渗硅后,液相硅与炭发生反应:C+Si→SiC。因此,在已知P0、P、ρ0和 ρ的情况下,假设反应前后体积不变,根据式(1)~(4),可计算出硅化SiC陶瓷各相含量。

式中VSiC、VSi和VC分别是硅化后SiC陶瓷中SiC、残余Si和未反应C相体积分数;P0和P分别是硅化前后制品开气孔率;ρ0和 ρ分别是硅化前后制品密度;1.99、2.33 和 3.22 分别是 C、Si和 SiC 相密度[9];12、28和40分别是C、Si和SiC相摩尔质量;mSi是参与反应的液相Si质量。
需要说明的是,公式中制品体积假设为1,制品单位体积下质量即为密度,硅化前后质量变化(ρ-ρ0)为残余硅质量与已参与反应的硅质量之和。与此同时,在假设反应前后制品体积不变的情况下,孔隙率的降低(P0-P)来源于残余硅与反应生成SiC引起的体积增加,以及炭反应引起的体积降低。
因此,为了获得硅化后SiC陶瓷各相含量,在已知硅化前密度和开气孔率的情况下(近似为1 300℃预制体密度和开气孔率),本试验对生成的硅化SiC陶瓷进行了密度和开气孔率测定。不同渗硅温度、保温60 min的SiC密度和开气孔率见图3。
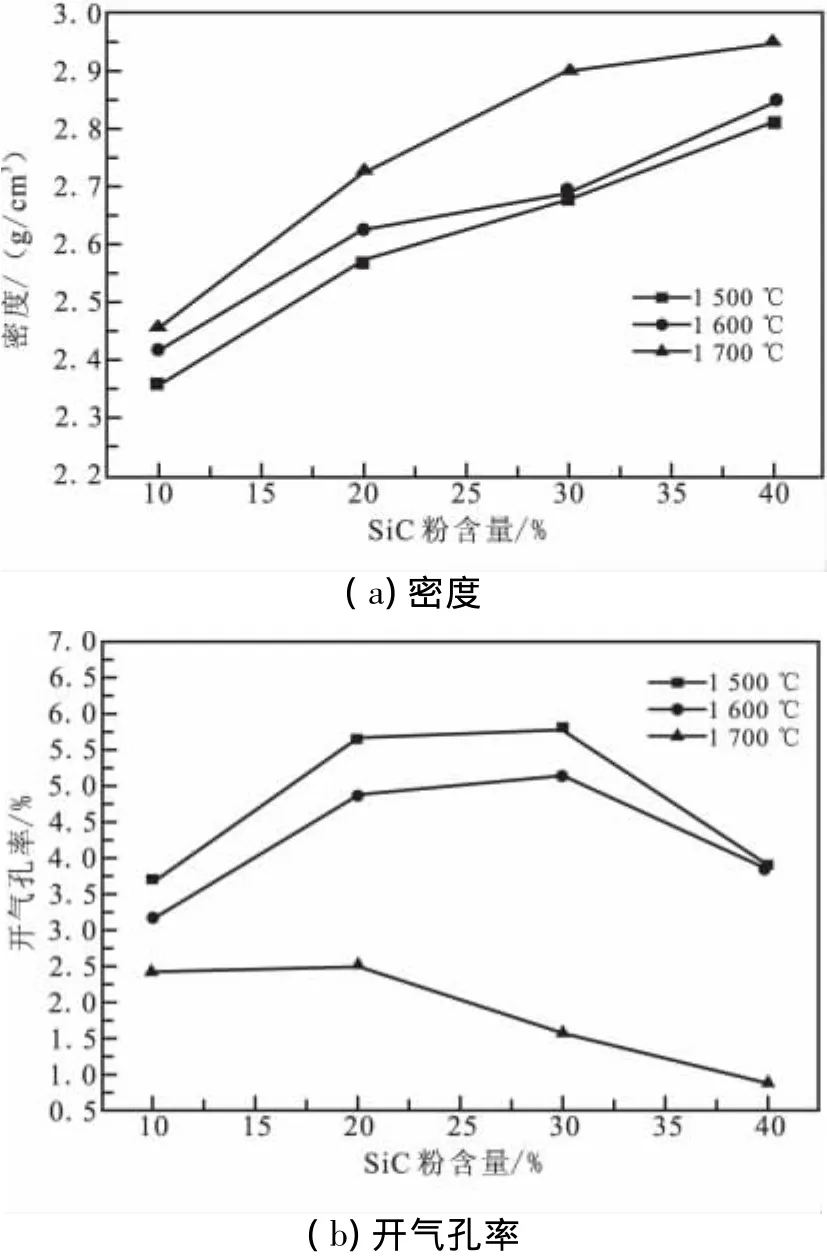
图3 硅化后制品密度及开气孔率随SiC粉含量的变化Fig.3 Variation of density and open porosity of siliconized sam ples w ith SiC powder content
由图3可看出,随原料中SiC粉掺量的增加,SiC陶瓷密度升高,这与预制体开气孔率有关。当渗硅前预制体开气孔率大时,就会有更多的液相硅渗入孔洞,并与炭反应生成SiC。与此同时,随渗硅温度升高,SiC陶瓷密度升高,开气孔率降低。然而,本试验测定的开气孔率随SiC粉掺量的增加呈现先增加后降低的变化规律。
2.2.1 渗硅温度和SiC粉掺量对各相含量的影响
本试验采用式(1)~(4)计算获得了不同渗硅温度下保温60 min后SiC陶瓷各相含量,其结果见图4。整体来说,随SiC粉掺量的增加,Si和SiC相体积分数提高,而C相体积分数降低。值得注意的是,当SiC粉掺量小于等于20%时,不同渗硅温度下残硅量均小于3.5%,而当 SiC粉掺量达到40%时,残硅量也小于13%。由此可见,以MCMBs为炭源有助于降低反应形成SiC的残硅量,从而可进一步提高制品性能,拓宽其使用范围。
由图4可见,随渗硅温度的提高,残硅量并没有显著的、有规律的变化趋势,残余炭相含量略有下降,而SiC相含量相对具有明显的增加。由此可见,渗硅温度的提高(1 500~1 700℃)除了可减小气孔率和提高制品密度外,对SiC陶瓷中SiC相含量影响最大。

图4 不同渗硅温度、保温60 m in制备的SiC陶瓷中Si、SiC和C相体积分数Fig.4 Volume fractions of Si,SiC and C phase in SiC ceram ics siliconized at different tem perature for insulation 60 m in
2.2.2 保温时间对各相含量的影响
图5示出了1 500℃渗硅,保温0、30、60 min的硅化SiC20RFSC制品各相含量变化趋势。通过测试,1 500℃渗硅,保温0、30 min后制品密度和开气孔率为2.43、2.55 g/cm3和4.82%、6.03%(保温60min 后制品密度和开气孔率见图3)。通过计算,未保温时残硅量10.06%,SiC 量为40.79%,残炭量为44.33%,而当保温30、60 min时,残硅量分别为1.62%、1.45%,SiC量分别为 54.84%、55.89%,残炭量分别为37.51%、37.00%。可看出,未保温时液相硅与炭之间虽然发生了化学反应,但反应还不够充分,制品内残留了较多的硅相和炭相。而当保温30 min以上时,硅化后制品各相含量没有太大差异,且残硅量很少,说明保温30 min便可使硅与炭充分反应。
据文献[4]报道,制备反应形成SiC的过程中,随渗硅时间的延长,会发生以下现象:(1)液相硅的快速浸渍;(2)固相炭快速溶解于液相硅中;(3)SiC的快速形成;(4)硅-炭界面形成连续SiC层;(5)炭通过SiC层扩散并与液相硅反应。一般情况下,前3种现象在1 min内即可发生,而形成最厚SiC层(约10μm)也仅需10~30 min。在随后的时间里,由于扩散控制的反应过程缓慢,因此渗硅30 min后各相含量没有太大差异。

图5 1 500℃渗硅、保温不同时间制备的20RFSC中各相体积分数Fig.5 Volume fractions of different phase in 20RFSC ceram ic siliconized at 1 500℃for different insulation time
2.3 XRD测试结果
图6为1 500℃渗硅制备的SiC陶瓷XRD测试结果,图中曲线(a)~(d)分别为1 500℃渗硅、保温1 h制备的10RFSC、20RFSC、30RFSC 和 40RFSC,(e)、(f)分别为保温0、30 min制备的20RFSC。从图6可看出,制品由β-SiC、C和Si相组成,按照衍射峰强弱依次是 β-SiC、Si、C相。由图6中(a)~(d)得出,随 SiC 粉掺量增加,β-SiC相及Si相衍射峰明显增强,但C相衍射峰没有显著变化规律,同时40RFSC制品无C相衍射峰,即C含量很少,以上结果与计算结果基本相符。从图6中(e)得出,虽然在1 500℃未保温,但制品主相依然是β-SiC相,同时C相衍射峰相对较强,说明残炭量较大。图6中(f)与(b)比较,各相衍射峰强度相近,表明各相含量相近。
2.4 微观结构
1 600℃渗硅制备的反应形成SiC断口抛光扫描照片见图7,而1 500℃渗硅制备的反应形成SiC断口抛光扫描照片与图7相似。黑色、灰色和白色区域分别为炭、SiC和残余硅。由图7可见,随SiC粉掺量的增加,炭体积分数降低,SiC和硅体积分数增加。这与计算结果和XRD结果吻合。在10RFSC表面,存在大量炭组分,其中包括一部分炭球。20RFSC表面形成网络状SiC,同时少量炭包含在SiC内部,残余硅几乎看不到。当SiC粉掺量小于等于20%时,MCMBs发生了较大的收缩,预制体开气孔率小于31%(见图1),液相硅的渗入受到阻滞,部分未反应炭保留于制品内。而SiC粉掺量大于等于30%,MCMBs收缩受到较大的抑制,使得预制体内具有较大的孔径和孔隙率(>36%),液相硅渗入量较大,深度较深。在30RFSC和40RFSC表面,主要组分为SiC相。值得注意的是,虽然有残余硅存在,但其分布比较均匀(图7(d))。

图6 1 500℃渗硅制备的SiC陶瓷XRD测试结果Fig.6 XRD patterns of SiC ceram ics siliconized at 1 500 ℃

图7 1 600℃硅化、保温60 m in制备的SiC陶瓷断口抛光扫描照片Fig.7 Polished cross-sections SEM images of reaction-formed SiC ceram ics siliconized at 1 600℃for insulation 60 m in
以上形成的特殊结构与MCMBs结构有密切关系。MCMBs在焙烧过程中,原有的炭层结构趋于平整和细化,在炭球内部与炭球之间形成许多通道,该现象是其他炭源没有的特征[10]。当熔融硅渗入时,就会充分进入这些通道形成网络状SiC。制品内孔隙的形成原因有:(1)预制体内的部分开孔被生成的 SiC堵死,Si的渗入被中止,就会形成封闭的孔;(2)部分炭球内层片间孔隙为闭孔,液相硅无法渗入;(3)未被硅化的炭球内层片在热处理过程中不断收缩,引起孔隙。
对SiC制品进行化学腐蚀,采用高倍SEM观察可以看到(图8),SiC陶瓷大部分组成是纳米级颗粒,同时存在部分微米级颗粒,说明本试验制备的SiC存在不同的硅/炭反应机理。据文献[7]报道,未经热处理的MCMBs中含有相当数量的小分子组分,使得大缩合芳烃之间的间隙被填充。随热处理温度的提高,MCMBs内部的缩合芳烃分子进一步发生缩合,形成更大的缩合分子,同时分子的平整度增加,在热处理过程中小分子组分变成气体从球体中逸出。进一步的升温过程中,大部分非炭杂原子从片状炭分子上脱离,并以气体形式随小分子逸出,造成炭层结构细化。这种细化后的结构可认为是无数非常细小的小颗粒定向排列组成炭微球,同时在小颗粒之间存在纳米级孔洞。另一方面,掺杂的纳米SiC填充于炭球之间,抑制炭球的收缩,在炭球之间形成微米级孔洞,即本试验制备的炭预制体存在双级孔。当熔融硅进入预制体内后,能够迅速进入双级孔。在微米级孔内,硅/炭反应机理主要为溶解-沉淀及扩散控制的反应机理,即熔融硅与炭发生反应生成SiC,新生成的SiC将原有SiC包裹,后续过程中炭通过SiC层扩散并与液硅反应,导致SiC颗粒不断生长,这部分颗粒尺寸较大。而在炭球内部,由于颗粒非常细小,熔融硅会立刻将其溶解并同时反应析出SiC,生成的SiC遗传了小颗粒定向排列的结构。从图8看出,腐蚀后SiC结构细小(约100 nm),大部分颗粒横向排列。

图8 SiC陶瓷腐蚀后断口形貌Fig.8 Fracture feature of SiC ceram ics after etching
此外,本试验制备的SiC表面未发现大的硅“湖”或“网络”。根据文献[5]报道,残余硅“湖”或“网络”的形成主要是Si+C放热反应产生的热膨胀应力引起。在MCMBs预制体中,炭球内层片之间的通道有助于释放热应力。因此,当渗硅温度在1 600℃以下时,没有发现大的残余硅“湖”或“网络”。同时高度有序的炭层结构,也有利于硅/炭反应的充分进行。
1 700℃渗硅制备的反应形成SiC颗粒整体变大,约十几微米,且残余硅不再是均匀分布,而在局部存在硅“网络”(见图9)。其原因是1 700℃硅化过程温度高、总的反应时间长,小颗粒SiC不断消融、合并、长大[11-12]。同时1 700℃时液相硅粘度最低,浸渍硅量最大,易形成硅“湖”或“网络”。此外,对于较高的渗硅温度,在保温结束后的自然降温时,易产生大的热应力,导致SiC颗粒间出现裂纹,使得部分液相硅进入。

图9 1 700℃渗硅、保温1 h制备的40RFSC断口抛光扫描照片Fig.9 SEM image of the polished cross-section of 40RFSC siliconized at 1 700℃for insulation 1 h
2.5 电阻率和强度
硅化后制品电阻率、抗弯强度随着SiC粉含量的变化如图10所示。

图10 硅化后制品电阻率、抗弯强度随SiC粉含量的变化Fig.10 Variation of resistivity and bending strength of siliconized samp leswith SiC powder content
本试验制备的反应形成SiC陶瓷具有较低的电阻率(图10(a)),尤其是1 700℃渗硅后制备的SiC陶瓷电阻率很低(SiC掺量由10%增至40%时,电阻率分别是1.09、1.31、0.49、0.36 μΩ·m),适应于其在众多领域的应用。SiC块体电阻率主要由制品孔隙率、组成相电阻率、相含量及相界面等决定。当渗硅温度一定时,随SiC粉掺量的增加,制品中SiC相含量升高,导致电阻率下降。但试验测得的电阻率变化趋势与开气孔率变化趋势相近,即先增加后降低的趋势,说明开气孔率是影响制品电阻率的主导因素。当渗硅温度由1 500℃升至1 700℃后,电阻率明显降低。其原因主要是渗硅温度提高后制品开气孔率降低,SiC相含量升高,以及SiC颗粒尺寸变大。颗粒粒径变大后,制品内相界面数量变少,电子碰撞时平均自由程变长,电阻率相应的降低。
陶瓷块体材料抗弯强度主要由颗粒尺寸、气孔率和其他缺陷等决定。由图10(b)可知,1 500、1 600℃渗硅制备的SiC陶瓷抗弯强度没有太大差异,其中1 600℃渗硅制备的 40RFSC抗弯强度最高,为359 MPa。此时虽然颗粒尺寸变大,但孔隙率降低,同时残余硅量少且均匀分布。而1 700℃渗硅制备的SiC陶瓷抗弯强度明显下降。尽管在该温度下获得的SiC陶瓷相相对较高,同时开气孔隙率较低,但由于此时陶瓷相颗粒尺寸较大,且局部出现硅“网络”,从而使得抗弯强度大幅下降。
3 结论
(1)随SiC粉掺量的增加,硅化后制品Si和SiC相体积分数提高,C相体积分数降低。渗硅温度提高可减小制品开气孔率和提高密度,同时SiC相含量相对明显提高。
(2)渗硅温度1 500℃、保温30 min以上时,硅与炭充分反应。
(3)当SiC粉掺量低时,MCMBs特殊结构有助于形成网络状SiC。而当SiC粉掺量达到40%时,制品主要成分为SiC,同时残余硅量少且均匀分布。但对于1 700℃渗硅制备的陶瓷制品,SiC颗粒较大,同时出现了残余硅“网络”。
(4)本试验制备的SiC制品具有较低的电阻率(最低值0.36μΩ·m)和较高的强度(最高值359 MPa)。渗硅温度为1 700℃时,制品电阻率最低,但抗弯强度大幅度下降。
[1] Chiang Y M,Messner R P,et al.Reaction-formed silicon carbide[J].Mater.Sci.Eng.A,1991,144(1-2):63-74.
[2] Singh M,Behrendt D R.Microstructure and mechanical properties of reaction-formed silicon carbide(RFSC)ceramics[J].Mater.Sci.Eng.A,1994,187(2):183-187.
[3] Calderon N R,Martínez-Escandell M,Narciso J,et al.The combined effect of porosity and reactivity of the carbon preforms on the properties of SiC produced by reactive infiltration with liquid Si[J].Carbon,2009,47:2200-2210.
[4] Margiotta JC,Zhang D,Nagle DC,etal.Microstructuralevolution during silicon carbide(SiC)formation by liquid silicon infiltration using optical microscopy[J].Int.J.Refract.Met.Hard Mater,2010,28:191-197.
[5] Wang Y,Tan S,etal.The effect of porous carbon preform and the infiltration process on the properties of reaction-formed SiC[J].J.Eur.Ceram.Soc.,2009,29:3091-3097.
[6] 许斌,陈鹏.中间相碳微珠(MCMB)的开发、性质和应用[J].新型炭材料,1996,11(3):4-8.
[7] 李同起,王成扬,刘秀军,等.中间相炭微球之炭层结构随温度演变的扫描电子显微镜分析[J].科学通报,2004,49(7):633-638.
[8] 倪红军,李飞,汤东,等.SiC掺杂对中间相碳微球组织的影响[J].上海交通大学学报,2008,42(12):2047-2051.
[9] Norfolk C,Mukasyan A,Hayes D,et al.Processing ofmesocarbonmicrobeads to high-performancematerials:Part II.Reaction bonding by in situ silicon carbide and nitride formation[J].Carbon,2006,44:293-300.
[10] Xia H,Wang J,Jin H,etal.Fabrication and properties of reaction-formed SiC by infiltrating molten Si into mesocarbon microbeads-based carbon perform[J].Mater.Sci.Eng.A,2010,528:283-287.
[11] 蔡宁,马荣,乔冠军,等.木材陶瓷化反机理的研究[J].无机材料学报,2001,16(4):763-768.
[12] 李世斌,宋士华,金志浩.硅/SiC高温热处理中组织及相组分变化[J].材料热处理学报,2006,27(2):11-14.
猜你喜欢
西部资源(2021年3期)2021-12-20 21:23:47
食品安全导刊(2021年21期)2021-08-30 08:21:24
科技创新导报(2019年36期)2019-05-06 09:08:14
中成药(2017年6期)2017-06-13 07:30:35
材料科学与工程学报(2016年4期)2017-01-15 13:35:29
陶瓷学报(2015年4期)2015-12-17 12:45:02
焊接(2015年6期)2015-07-18 11:02:24
汽车文摘(2014年9期)2014-12-13 13:10:30
中成药(2014年7期)2014-02-28 22:28:05
长江大学学报(自科版)(2013年1期)2013-10-26 09:08:47
