
海洋生物抗菌肽在枯草杆菌BS168中的高效表达
2011-03-14 06:06
海洋科学 2011年3期
(蚌埠学院 生物与食品工程系,安徽 蚌埠 233030)
马蹄蟹(Horseshoe crab)是一类远古的海洋节肢动物。马蹄蟹血细胞中分离提取的一类抗菌肽[1],具有抗细菌、抗真菌、抗病毒、抑制肿瘤细胞增殖和诱导癌细胞分化的生物活性[2]。抗菌肽tachyplesinⅠ因相对分子质量小不具备抗原性,对原核生物细胞有很高的杀菌活性,对真核生物的正常细胞则无明显毒副作用。抗菌时一般没有特殊受体,不会诱导抗药菌株的产生,是一类具有巨大潜在应用价值的抗菌肽。但由于马蹄蟹是一种珍贵的海洋生物,物种稀少,是濒临灭绝生物,且 tachyplesinⅠ的分离纯化过程繁琐且费用较高;通过化学合成制备,价格昂贵,生物活性低;因此采用基因工程制备tachyplesinⅠ成为一种可行的方案[3]。
目前,利用基因重组技术在体外大量制备活性多肽是生物制品研究的热点之一。对于分子质量较小的多肽,如果用常规方法构建单拷贝基因的重组质粒,则目的基因在细胞中的表达量很低,无法体现基因工程产量高的优势。因此,本文建立了一种构建串联基因的方法,以实现小分子多肽在体外的高效表达,为小肽的串联表达提供了理论和实践依据。本研究为了克服基因工程制备 tachyplesinⅠ的缺陷,设计构建 tachyplesinⅠ基因串联表达载体,并将重组基因成功转化到分泌型表达系统枯草杆菌 BS168中,实现了抗菌肽tachyplesinⅠ的高效分泌表达。
1 材料与方法
1.1 材 料
1.1.1 菌株和质粒
克隆宿主菌Escherichia coli.DH5α、大肠杆菌E.coliK88、金黄色葡萄球菌(Staphyloccocus aureus)为本室保存;枯草杆菌Bacillus subtilisBS168为表达宿主菌,由上海生命科学院植物生理与生态研究所惠赠;TA克隆载体 pUCM-T购自上海生工公司;大肠杆菌-枯草杆菌穿梭质粒 pSBPTQ[4]由中山大学罗进贤教授惠赠。
1.1.2 培养基和主要试剂
马蹄蟹购自广东省深圳市水产品市场;枯草杆菌感受态制备用GMI、GMII培养基[5];发酵用MSR培养基[6]。cDNA合成试剂盒及各种工具酶均购自TaKaRa公司;Trizol reagent购自 Invitrogen公司;蛋白分子质量标准、mRNA纯化试剂盒购自Promega公司;CM Sepharose购自Amersham Biosciences;其他生化试剂(分析纯)购自上海生工;实验中所用引物(由上海生工合成),各引物设计如表1所示。

表1 所用引物表Tab.1 PCR primers
1.2 方法
1.2.1 抗菌肽tachyplesinsⅠ基因的RT-PCR扩增
从马蹄蟹围心腔采集血液,8 000 r/min 离心10 min 获得血细胞[7],用 Trizol处理提取总 RNA,以 mRNA 纯化试剂盒获得 mRNA。反转录合成cDNA,进一步以cDNA 为模板,参考Genbank收录的 tachyplesinⅠ基因序列,设计了上下游引物Tac-1和Tac-2,见表1。并设计了XbaⅠ和KpnⅠ酶切位点和保护碱基。PCR扩增获得的目的基因tac,经2.5%琼脂糖凝胶电泳分析,电泳检测后测序。
1.2.2 串联基因(2tac)的设计
参照 Genbank上公布的基因序列,设计合成两条完全互补单链 tachyplesinsⅠ串联基因,5’端设计XbaⅠ黏性末端,3’端设计KpnⅠ黏性末端。通过上海生工公司合成带有上述黏性末端的完全互补的碱基序列,退火合成带有黏性末端的双链串联tachyplesinsⅠ基因。方法合成如下:取等量摩尔浓度25 μmol/L的正反义引物各 4 μL,添加超纯水至40 μL。退火条件:80℃水浴退火5 min,然后自然冷却至30℃,其过程大概3 h。退火合成串联基因(2tac)克隆连接到pUCM-T上进行初步鉴定及目的串联基因的扩增。

1.2.3 串联基因2tac的克隆鉴定及体外PCR扩增
克隆载体 pUCM-T经限制性内切酶XbaⅠ和KpnⅠ双酶切后,和tachyplesinⅠ基因tac及 2tac经T4 DNA 连接酶连接。转入大肠杆菌DH5α培养,经过蓝白斑筛选得阳性克隆,培养提取质粒。经双酶切初步鉴定,再通过测序进一步验证克隆片断的正确性。所得克隆质粒命名为 pUCM-2TAC和 pUCMTAC。
1.2.4 表达载体的构建及鉴定和重组质粒转化
以 pUCM-2TAC质粒模板,通过引物 M13经PCR扩增得到串联基因2tac,PCR反应条件:94℃预变性10 min;94℃变性30 s,55℃退火30 s,72℃延伸30 s,共进行35个循环;最后72℃延伸10 min。将PCR产物和表达载体 pSBPTQ同时进行XbaⅠ和KpnⅠ双酶切,T4 DNA连接酶连接,大肠杆菌转化按Sambrook的方法进行[8]。转化后筛选阳性克隆,双酶切鉴定,测序验证。所得质粒命名为 pSBPTQ-2TAC,重组表达质粒 pSBPTQ-TAC和 pSBPTQ-2TAC转化枯草杆菌BS168的方法参照Harwood[9]方法进行。
1.2.5 诱导表达及SDS-PAGE电泳分析
挑取新鲜转化的枯草杆菌工程菌BS168/pSBPTQ-TAC和 BS168/pSBPTQ-2TAC单菌落接种于含卡那霉素(10 mg/L)的LB培养基中,37 ℃ 250 r/min培养过夜,作为种子液。然后种子液按5 %接种量转接于含卡那霉素(10 mg/ L)的发酵培养基(MSR)中,37℃ 250 r/ min 振荡培养2~4 h,加蔗糖至终浓度2 %诱导表达[10],每隔 12 h依次取样,收集的发酵液经10 000 r/min离心 15 min,取发酵液上清进行Tris-Tricine SDS-PAGE(16.5%)电泳检测,以确定最佳诱导时间。用含质粒 pSBPTQ的对照菌体和标准蛋白作对照。
1.2.6 重组tachyplesinⅠ的分离纯化
收集发酵液12 000 r/min,离心15 min。向发酵上清液中依次加入硫酸铵使终浓度达到35%、45%、55%、65%、75%,12 000 r/min离心30 min,收集沉淀,用pH7.4的Tris-HCl缓冲液溶解沉淀。将盐析浓缩效果最好的溶液装入透析袋中进行透析。透析产物上CM -Sepharose层析柱,用10 mmol/L Tris-HCl(pH6.8)平衡洗脱柱,然后用相同缓冲溶液配制的0~1.0 mol/L的NaCl线性梯度洗脱(2 mL/min),收集活性峰,对收集样品经冷冻干燥浓缩,用无菌水配得 tachyplesinsⅠ表达产物溶液 10 mL。参照谢海伟等[11]高效液相色谱方法检测 tachyplesinⅠ表达产物含量。
1.2.7 TachyplesinⅠ串联表达产物2TAC的水解及活性检测
取两份各为 1.0 mL冷冻干燥的串联表达产物2TAC,将其中一份溶于 1.0 mL BrCN溶液(50 g/L,70 %甲酸配制)中,另一份溶于仅含70 %甲酸溶液中,同时置于 4 ℃冰箱水解处理 24 h,水解后再加1.0 mL的双蒸 H2O,冷冻干燥以除去甲酸和 BrCN,干制品用无菌水重新配制成1.0 mL溶液进行活性检测。上述2TAC水解液产物进行活性鉴定,以大肠杆菌 K88和金黄色葡萄球菌为试验菌株,分别取5 mg/L的2TAC和TAC,10 mg/L的2TAC和2TAC水解产物,以 BS168/pSBPTQ空质粒菌株发酵液为对照,参照微量肉汤稀释法[12]进行抗菌活性检测。同时以上述TAC对供试菌株孵育,参照代建国等[13]建立方法进行透射电镜观察,分析细胞微观结构的变化。
2 结果与分析
2.1 TachyplesinⅠ串联基因(2tac)的设计
TachyplesinⅠ串联基因的设计如图1所示,在单 tachyplesinⅠ基因tac的 3’端添加“AAC”密码子,目的为了使 tachyplesinⅠ基因表达产物 C末端产生酰氨化结构,两个 tachyplesinⅠ基因通过蛋氨酸密码子“ATG”连接,目的在于增添表达产物的BrCN水解位点。为了便于和表达载体连接,在串联基因的5’端设计XbaⅠ酶切位点黏性末端,后加两个“AA”为使串联基因 2tac在表达载体的正确阅读框内,使其能正确表达,其前加“AA”作为保护碱基。
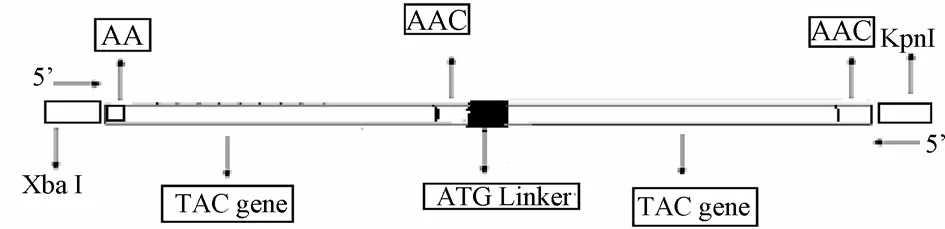
图1 tachyplesinⅠ串联基因2tac的设计Fig.1 Design of tandem gene of tachyplesinⅠ
2.2 串联基因2tac重组表达载体的构建
将大肠杆菌质粒 pSP72的复制起点及氨苄青霉素抗性基因转移到枯草杆菌质粒pUB18上构建了大肠杆菌-枯草杆菌穿梭型克隆载体 pSB。然后将枯草杆菌sacB基因的启动子-信号肽序列(sacB P.S.)及地衣芽孢杆菌α-淀粉酶基因的转录终止信号T(amyT)克隆进 pSB,获得诱导型表达-分泌载体 pSBPT,最后将短小芽孢杆菌正调控基因degQ引入pSBPT,获得重组质粒pSBPTQ,构建过程见图2。含2tac基因的重组质粒的构建如图3所示,将pSBPTQ用XbaⅠ/KpnⅠ双酶切,电泳回收7.5 kb大片段,与经XbaⅠ /KpnⅠ酶切的 PCR产物连接,构建含2tac基因的重组质粒。
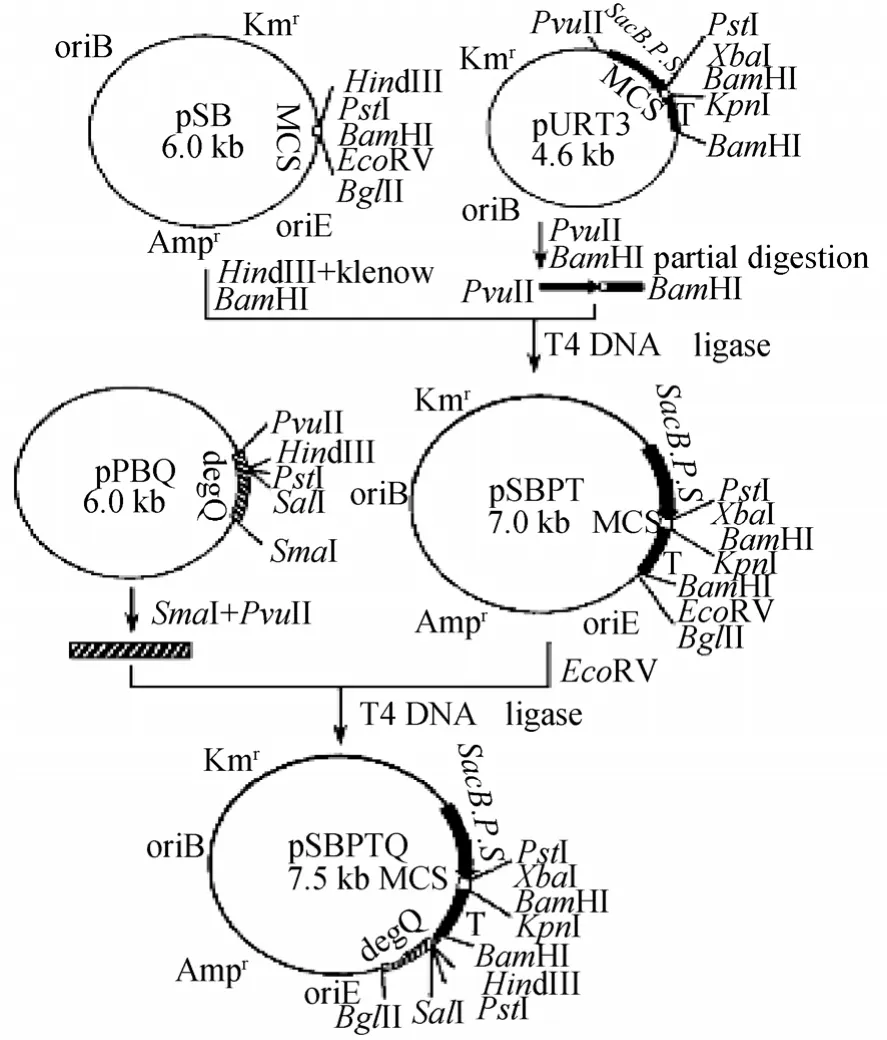
图2 穿梭型诱导表达载体构建 [4]Fig.2 The expression plasmid pSBPTQ
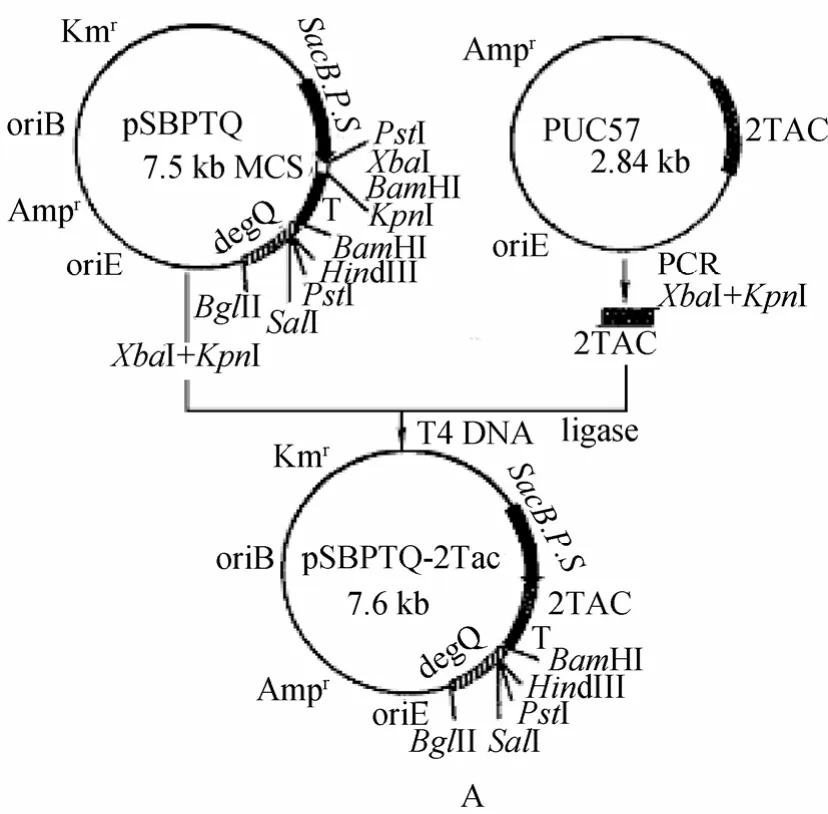
图3 重组表达载体pSBPTQ-2TAC的构建Fig.3 The recombinant expression plasmid pSBPTQ-2TAC
2.3 TachyplesinⅠ基因(tac)的 RT-PCR 扩增以及电泳鉴定
从马蹄蟹细胞中提取总 RNA,根据tachyplesinⅠ基因序列设计的引物Tac-1和Tac-2,以马蹄蟹血细胞cDNA为模板扩增的PCR产物,产物经凝胶电泳分析,如图4所示。在泳道2中出现一条70 bp左右的DNA 条带。采用退火法合成的串联基因2tac,经凝胶电泳分析,在泳道1中出现一条120 bp左右DNA条带。初步证明获取了目的基因tac和2tac。
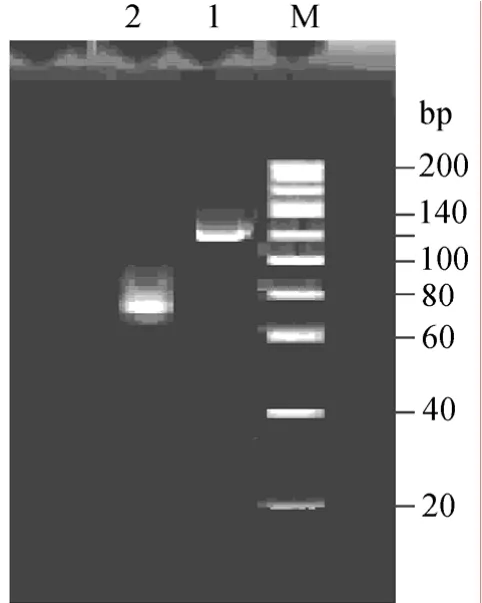
图4 tachyplesinⅠ基因tac和2tac 的PCR产物电泳图Fig.4 PCR product of tac gene and 2tac
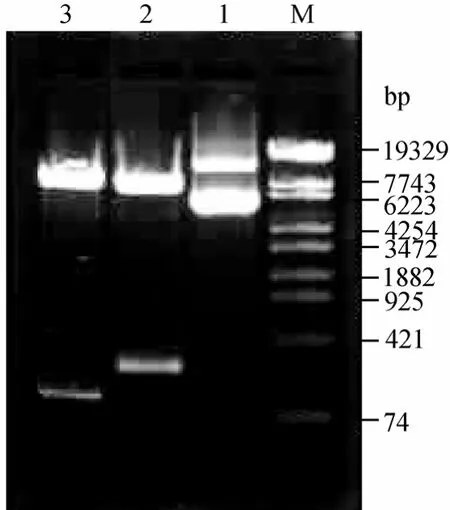
图5 重组子pSBPTQ-TAC和pSBPTQ-2TAC的酶切鉴定Fig.5 Restriction enzyme analysis of pSBPTQ-TAC and pSBPTQ-2TAC
2.4 重组表达载体的鉴定
重组质粒经过双酶切后凝胶电泳图见图5,结果显示在约80 bp、150 bp和7.4 kb处有预期的条带。初步证明目的基因tac和2tac已经克隆到表达载体上。重组子进行测序,测序图谱如图6,7所示,测序结果采用软件 CLUSTAL(1.81)进行克隆序列和已知tachyplesinⅠ基因序列进行比对分析。核苷酸序列比对结果,重组子基因tac和 2tac核苷酸序列和已知tachyplesinⅠ基因序列完全相同,长度分为 56 bp、119 bp,这表明tachyplesinⅠ基因tac和2tac成功克隆到pSBPTQ载体中,通过测序图谱进一步表明,目的基因连接顺序和启动子方向一致,在表达载体的正确阅读框内,重组子命名为 pSBPTQ-2TAC和pSBPTQ-TAC。
2.5 重组转化菌的诱导表达及 SDS-PAGE鉴定
与对照 BS168/pSBPTQ发酵液相比,在重组子BS168/pSBPTQ-2TAC和BS168/pSBPTQ-TAC的电泳图中出现一条明显的蛋白条带,如图8中箭头所指条带分别大小约为2.0 KD和5.0 KD,表达产物大小与天然 tachyplesinⅠ多肽和其串联体的相对分子质量大小相符,由此可以初步确认目的蛋白 2TAC和 TAC在宿主菌种得到了有效地表达,并且分泌到发酵液中。采用凝胶成像系统分析软件 GeneTools对SDS-PAGE电泳的结果进行分析,对图8中2,3泳道进行扫描,经峰面积积分计算出目的条带的吸收峰在所有表达蛋白的吸收峰中所占的百分含量,即为目的蛋白在表达的全蛋白中的百分含量。本实验比较的是TAC和2TAC蛋白在全蛋白中的百分含量,经GeneTools软件分析,重组表达产物TAC和2TAC蛋白表达量分别约为8.5%、15.8%。
工程菌株B.subtilisBS168/pSBPTQ-2TAC和BS168/pSBPTQ-TAC,经蔗糖诱导后,每隔 12 h取样进行 SDS-PAGE电泳检测。由图9可以看出,目的蛋白2TAC和TAC随着诱导时间的增加,表达量也在增加,当诱导24 h时,表达产量达到最高值。
2.6 表达蛋白2TAC和TAC的分离纯化及SDS-PAGE鉴定
通过不同饱和浓度的硫酸铵对表达产物进行盐析初步纯化,浓缩产物进行 MIC 检测,目的蛋白沉淀量与硫酸铵浓度的关系见图10所示,用55%饱和度的硫酸铵处理后,浓缩液抗菌活性最强,用55%饱和度的硫酸铵进行盐析可得到最大量的目的蛋白。因此,在分离纯化过程中,先用55%饱和度的硫酸铵沉淀目的蛋白,进行浓缩处理。
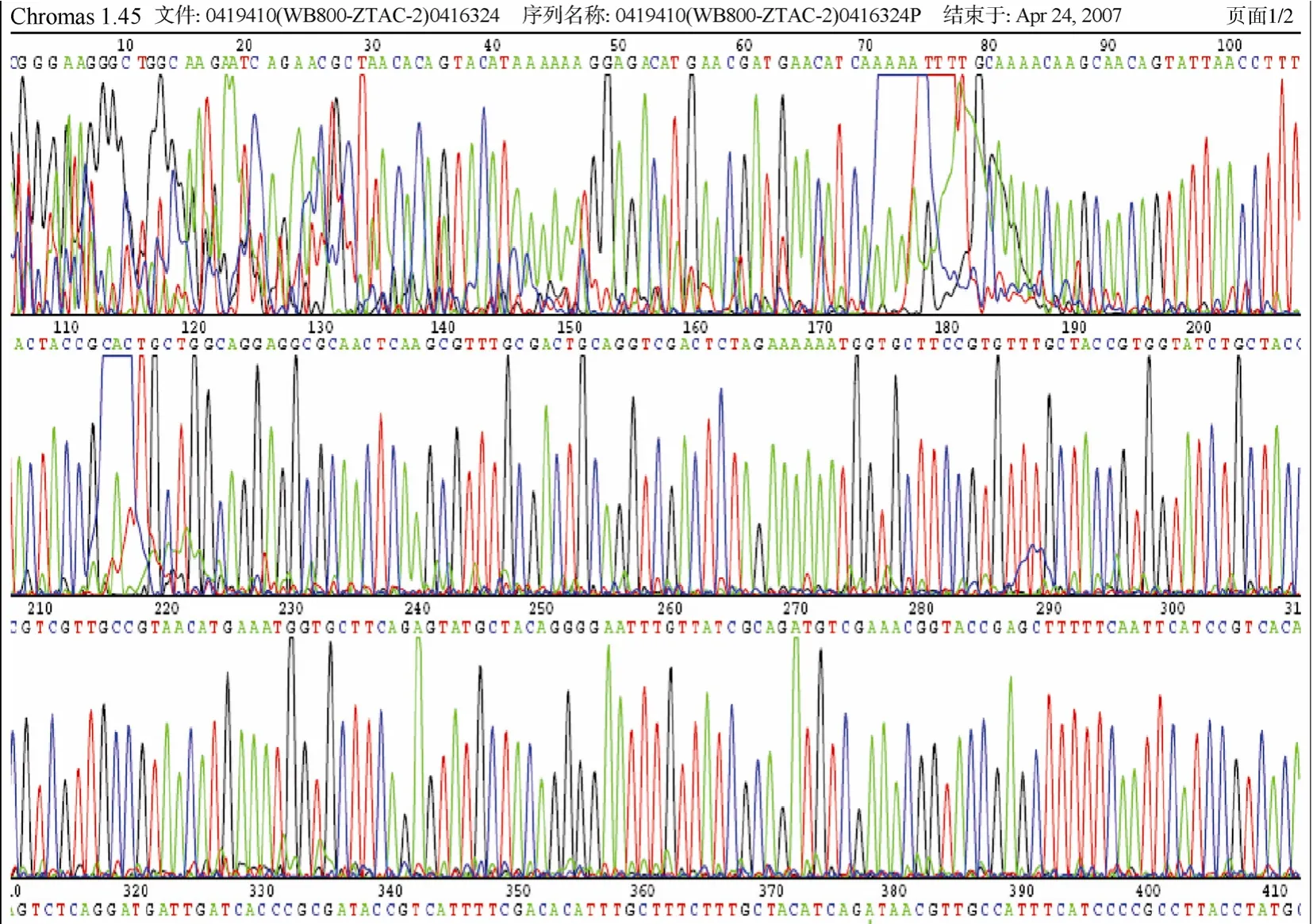
图6 重组表达质粒pSBPTQ-2TAC测序图谱Fig.6 DNA sequence of pSBPTQ-2TAC
图7 重组表达质粒pSBPTQ-TAC测序图谱Fig.7 DNA sequence of pSBPTQ-TAC
发酵液经 55%饱和度的硫酸铵盐析沉淀后,在蒸馏水中透析。透析液经CM-Sepharose Fast Flow柱层析,结果图12(A,B)中显示出现 4个蛋白峰,对各个峰进行抗菌活性分析后,第Ⅳ峰具有抗菌活性,如图12(A,B)中箭头所指。目的产物所在的第Ⅳ峰在低浓度0.1~0.2 mol/L左右的NaCl被洗脱下来,冷冻干燥,用无菌水配得 tachyplesinⅠ表达产物溶液10 mL。离子交换柱收集的表达产物,经高效液相色谱分析浓缩的表达产物TAC和2TAC的浓度分别为5.46 mg/L和10.36 mg/L。纯化后经过SDS-PAGE分析结果见图11,由电泳结果可见,重组的 2TAC和TAC表达产物经分离纯化后,获得均一的蛋白条带,分子质量大小和上述鉴定相符。表明重组的 2TAC和TAC表达产物获得成功。
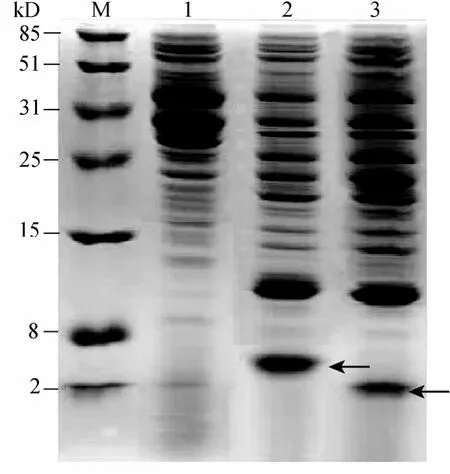
图8 表达产物2TAC和TAC的SDS-PAGE分析Fig.8 SDS-PAGE analysis of recombinant protein
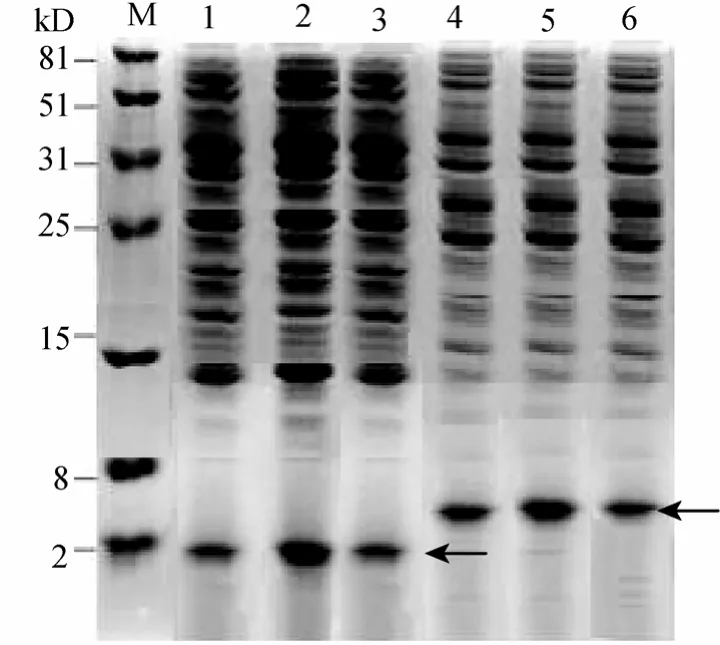
图9 不同诱导时间下表达产物 2TAC和 TAC的 SDSPAGE分析Fig.9 SDS-PAGE of 2TAC and TAC at different induced times
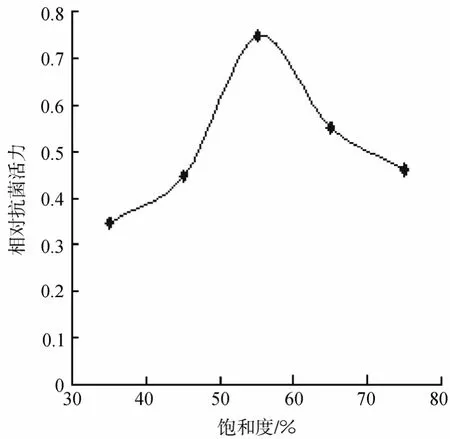
图10 发酵液在不同硫酸铵饱和度盐析下的相对活性Fig.10 The relative activity of fermentation in different concentrations of (NH4)2SO4
2.7 对发酵上清液进行了活性鉴定,抑菌活性鉴定
通过对表达产物TAC和2TAC以及2TAC的水解产物的抑菌圈观察,诱导表达的目的蛋白溶液对E.coliK88和S.aureus的抑菌试验结果见(图13)。从图13可以看出,tachyplesinⅠ串联表达产物2TAC的抑菌圈明显小于单拷贝表达产物 TAC的抑菌圈,而2TAC经BrCN水解后的抑菌圈又明显大于单拷贝表达产物 TAC的抑菌圈,这表明 tachyplesinⅠ串联体2TAC经水解后对大肠杆菌K88和S.aureus都具有明显的抑菌活性。
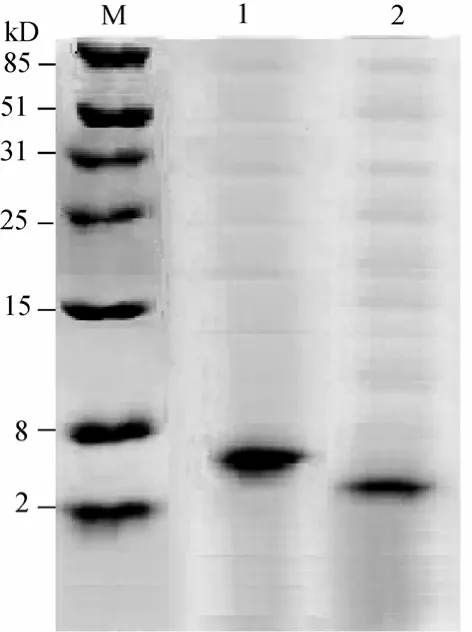
图11 纯化后2TAC和TAC的SDS-PAGE分析Fig.11 SDS-PAGE analysis of purified TAC and 2TAC
同时表达产物TAC和供试菌大肠杆菌K88,金黄色葡萄球菌(S.aureus)共同孵育后,从透射电镜观察(图14)发现,对照组(图14-A,C)细胞结构完整,细胞外观平滑,细胞壁膜完整,细胞饱满,细胞内容物充实、致密;处理组(图14-B,D)可以看出几点变化,部分细胞膜出现裂解,基本细胞形态保持,细胞外观粗糙,部分细胞壁膜裂解,处理后金黄色葡萄球菌有两处明显的细胞膜出孔道现象,细胞质结构松弛,出现少许绒毛;处理后大肠杆菌 K88细胞质内空,部分细胞器和细胞核区消失。上述透射电镜观察表明表达产物TAC具有抗菌活性。
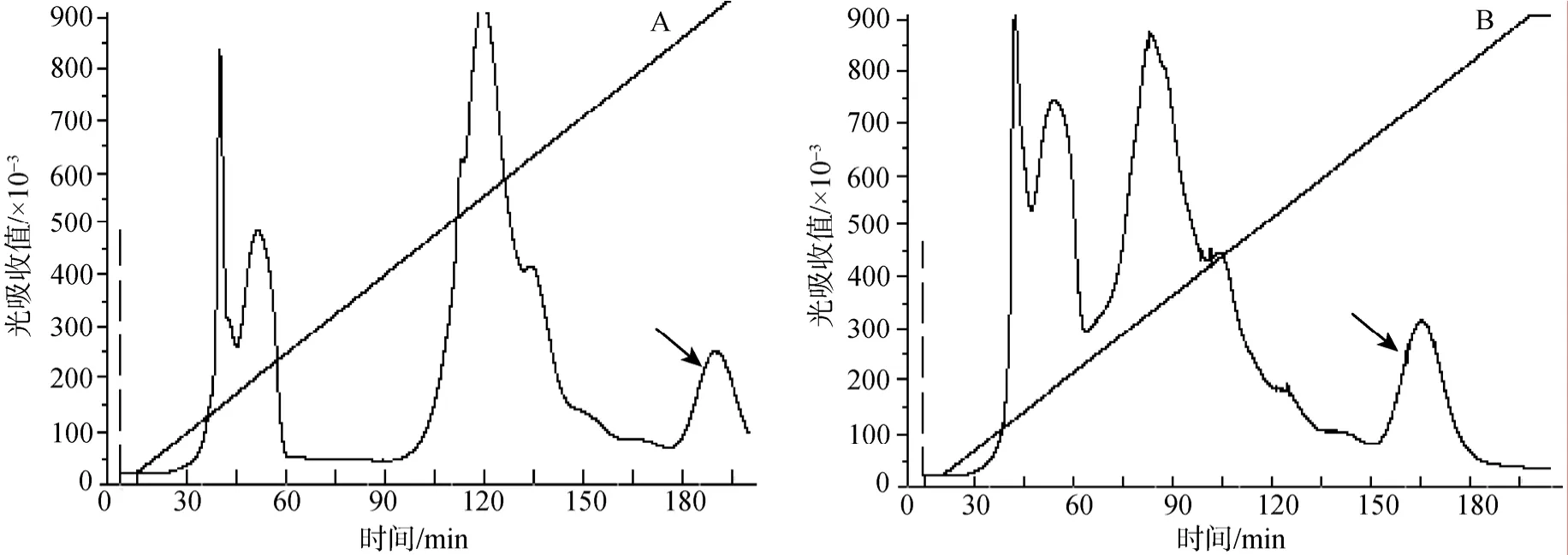
图12 CM-Sepharose Fast Flow阳离子交换层析图Fig.12 2TAC and TAC purified by CM-Sepharose
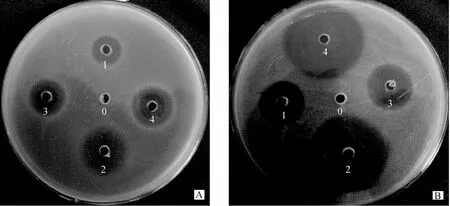
图13 TAC和2TAC及水解后2TAC对E.coli K88和S.aureus的抑菌圈
3 讨论
本实验通过前期对 tachyplesinⅠ抗菌谱的研究[10],发现抗菌肽 tachyplesinⅠ对枯草杆菌的抑制作用,明显低于对大肠杆菌的抑制作用,因此我们尝试利用枯草杆菌进行tachyplesinⅠ的表达。本实验首次完成了 tachyplesinⅠ在枯草杆菌表达系统的高效表达,在此之前进行的抗菌肽 tachyplesinⅠ或类似抗菌肽的基因表达研究中,只有通过大肠杆菌和酵母表达系统表达成功的,但表达产量都很低。如张春义[14]用大肠杆菌表达成功,对黄曲霉孢子具有活性;张钫等[15]成功表达出抗菌肽 polyphemusinⅡ,并具有抗菌活性。

图14 产物TAC处理的透射电镜图及未处理对照Fig.14 Transmission electron microscopy of S.aureus and E.coli K88 after treated by TAC
本实验首次用枯草杆菌表达系统进行tachyplesinⅠ串联表达,利用大肠杆菌-枯草杆菌穿梭表达载体pSBPTQ实现了串联基因2tac和单拷贝基因tac在枯草杆菌 BS168中高效的表达。从表达产物的生物活性可以看出,串联表达产物 2TAC具有抗菌活性,但其抗菌活性低于单拷贝表达产物TAC,当串联表达产物2TAC用BrCN水解后,水解液的抗菌活性明显增加,其最小抑菌浓度和单拷贝表达产物TAC相近。同时串联表达蛋白在连接处通过溴化氢水解后,可使表达产量增倍,这种方法有利于提高 tachyplesinⅠ表达产量,同时降低表达产物对宿主细胞的毒性,实现 tachyplesinⅠ的高效表达。这一研究结果为tachyplesinⅠ高效表达提供了重要研究方法,具有非常重要应用价值。
本实验采用枯草杆菌表达系统和串联基因表达方式,克服了表达产物对宿主的抑制作用,大大提高了产量,取得一定成功,但仍有许多地方需要改进:(1)本实验采用的宿主菌 BS168是枯草杆菌表达系统中的最为广泛应用宿主菌,有许多优点,并在本实验中表达成功,但宿主菌 BS168仍然存在一定问题,在BS168中已发现碱性蛋白酶、中性蛋白酶、金属蛋白酶、中性蛋白酶 B和三种丝氨酸蛋白酶等多胞外蛋白酶,当外源蛋白分泌表达后,容易被这些蛋白酶降解,从而大大降低产率。枯草杆菌的这种的特性,可能部分水解抗菌肽,造成表量低,在枯草杆菌 168宿主菌基础上通过蛋白酶基敲除等手段,已经发展到多种蛋白酶缺陷型的宿主菌,如 8种蛋白酶的缺陷型WB800是剔除8种蛋白水解酶的工程菌,因此可选择更理想的表达系统[16]。另外可以在本文基础上选择一些真菌表达系统,如酵母表达系统,霉菌表达系统或植物表达系统;(2)鉴于本实验 2拷贝的串联基因可以实现正确的表达,因此可以通过设计新的串联基因方法,实现多拷贝的串联基因进行表达,以进一步降低表达产物对宿主抑制,同时水解后成倍提高抗菌肽产量;(3)根据抗菌肽tachyplesinⅠ氨基酸序列特点,寻求更合理且无毒害的酶切位点;设计更为合理的,用无毒的,成本低的酶水解代替BrCN水解。
[1]Nakamuro T,Furunaka H,Miyata T, et al.Tachyplesin,a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus) [J]. J Biol Chem,1988,263(32):16 709-16 713.
[2]Morvan A,Iwanaga S,Comps M,et al.In vitro activity of the limulus antimicrobial peptide tachyplesin I on marine bivalve pathogens [J].J Invertebr Pathol,1997 ,69(2):177-182.
[3]谢海伟,代建国,郭勇.鲎源抗菌肽的研究及其潜在应用价值[J].生物技术通讯,2007,18(3):530-533.
[4]李瑞芳,张添元,罗进贤.双功能枯草杆菌诱导型高效表达分泌载体的构建与鉴定[J].微生物学报,2006,46 (5):714-719.
[5]Dubnau D,Davidoff A R.Fate of transforming DNA following uptake by competent Bacillus subtilis.I.Formation and properties of the donor-recipient complex[J].J Mol Biol,1971,56:209-221.
[6]Ye R,Kim J H,Kim B G, et al.High-level secretory production of intact,biologically active staphylokinase fromB.subtilis[J].Biotechnol Bioeng,1999,62(1):87-96.
[7]郑伟,韩文瑜,韩俊友,等.中国鲎(Tachypleus tridentatus)基因工程抗菌肽的制备及其抗菌活性[J].中国兽医学报,2007,27(2):211-214.
[8]Sambrook J,Fritsch E F,Maniatis T.Molecular Cloning:A Laboratory Mannual[M].New York:Cold Spring Harbor Laboratory,1989.
[9]Harwood C R,Cutting S M.Molecular biological methods for Bacillus [M].England:John Wiley &Sons,1990.
[10]Ye R Q,Kim J H,Kim B G, et al.High-level secretor production of intact,biologically active staphylokinase fromB.subtilis[J].Biotechnol Bioeng,1999,62(1):87-96.
[11]谢海伟,代建国,郭勇,等.鲎素抗菌肽的分子结构稳定性及生物活性华[J].华南理工大学学报(自然科学版),2008,36(4):144-150.
[12]Giacometti A,Cirioni O,Barch iesi F,et al.In vitro susceptibility tests for cationic peptides:comparison of broth microdilution methods for bacteria that grow aerobically[J].Antimicrob Agents Chemother,2000,44(6):1 694-1 696.
[13]代建国,谢海伟,金刚,等.鲎素的抗菌靶点初探[J].生物化学与生物物理进展.2008,35(5):563-569.
[14]张春义,范云六.抗真菌鲎素基因在大肠杆菌中的表达[J].农业生物技术学报,1998,6(3):211-216.
[15]张钫,郑伟,韩文瑜.鲎抗菌肽 polyphemusinⅡ基因的改造、克隆及其在大肠杆菌中的表达[J].新疆农业学报,2005,42:31-34.
[16]Wu S C,Yeung J C,Wong S L.Functional production and characterization of a fibrin-specific single-chain antibody fragment fromBacillus subtilis:effects of molecular chaperones and a wall-bound protease on antibody fragment production [J].Appl Environ.Microbiol,2002,68:3261-3269.
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13
新世纪智能(英语备考)(2018年11期)2018-12-29
天然产物研究与开发(2018年7期)2018-08-21
广东饲料(2016年5期)2016-12-01
中国环境监察(2016年7期)2016-10-23
中国现当代社会文化访谈录(2016年0期)2016-09-26
国外医药(抗生素分册)(2016年5期)2016-07-12
中学化学(2016年2期)2016-05-31
课程教育研究·下(2016年2期)2016-03-25
探测与控制学报(2015年4期)2015-12-15
