
小反刍兽疫病毒N基因的原核表达
2011-03-07 06:13:12秦峻岭高玉伟杨玉姣杨松涛王承宇王铁成夏咸柱
动物医学进展 2011年3期
阮 洋,秦峻岭,高玉伟,孙 玮,张 涛,4,杨玉姣,杨松涛,王承宇,王铁成,黄 耕,夏咸柱,3*
(1.吉林农业大学动物科学技术学院,吉林长春130118;2.军事医学科学院军事兽医研究所 吉林省人兽共患病预防与控制重点实验室,吉林长春130062;3.吉林大学畜牧兽医学院,吉林长春130062)
小反刍兽疫病毒(Peste des petits ruminants virus,PPRV)是危害山羊和绵羊的一种高度接触性病毒,其高致病率和死亡率对养羊业造成重大的经济损失。临床上早期表现出高热、眼炎、脓性鼻液、腐烂性口炎,后期表现为胃肠炎和肺炎。小反刍兽疫由于其高传染性和快速传播的能力被世界动物卫生组织(OIE)列为必须报告的动物疫病。小反刍兽疫的诊断主要靠免疫捕捉ELISA及病毒分离鉴定。近10年来,这些检测技术逐渐被基于染色体组的检测手段所替代。例如,在2005年由Forsyth M A等[1]建立的RT-PCR方法,在保守的P基因和F基因设计数对引物用于鉴别牛瘟病毒(RPV)和PPRV。PPRV N蛋白特异性单克隆抗体已经被用来诊断病毒抗原,N基因的cDNA已经作为探针用来诊断PPRV[2]。
N蛋白由N基因编码,基因总长度为1 689 nt。N基因开放阅读框长度为1 578 nt,N蛋白分子质量约为57.18 ku。包含一个开放阅读框(ORF),编码525个氨基酸残基。N蛋白紧紧地包裹着RNA,并形成螺旋结构,起到保护核酸的作用。N蛋白的抗原性稳定,在病毒感染的动物血清中针对N蛋白的抗体占主导地位,但是此类抗体不具有中和病毒的作用。N蛋白结构中分为4个区域,即Ⅰ区(氨基末端区)、Ⅱ区(123-144氨基酸)、Ⅲ区(蛋白的中部区域)、Ⅳ区(421-525的羧基末端区)。与其他麻疹病毒属成员相比,Ⅰ区和Ⅲ区保守性较高。用抗N蛋白的单克隆抗体可以区别RPV和PPRV毒株,从而进行鉴别诊断。PPRV N蛋白是组成病毒核衣壳的主要蛋白之一,Ismail T M等[3]克隆表达了N蛋白用于血清学诊断。Libeau G等[4-5]获得了抗N蛋白的单克隆抗体,并建立了竞争ELISA和免疫捕获ELISA方法用于PPR的血清学诊断,各国研究者先后建立了针对N基因的RT-PCR方法用于PPRV的病原检测。
我国在2007年暴发小反刍兽疫疫情[6],造成严重的经济损失。目前针对小反刍兽疫没有有效的治疗手段,主要靠疫苗进行防控。因此建立快速有效的检测方法就显得尤为重要。本试验对小反刍兽疫的N蛋白进行原核表达,旨在获得高纯度的蛋白抗原,为下一步小反刍兽疫基因工程疫苗的开发、单克隆抗体的制备及快速诊断方法的建立提供物质基础。
1 材料与方法
1.1 材料
原核表达载体pET-28a(+)、羊抗PPRV Nigeria75/1株多克隆抗体由本实验室保存;MEN培养基为Gibco产品;PMD-18Ts载体,各种限制性内切酶、DNA标准DL 15 000、DL 2 000、exTag、dNTP全部为宝生物工程(大连)有限公司产品;预染蛋白Marker宽范围(6~175),为NEB生物公司产品;鼠抗his标签单抗、辣根过氧化物酶标记的羊抗鼠IgG、辣根过氧化物酶标记的兔抗山羊IgG为北京中杉金桥生物技术有限公司产品。
1.2 方法
1.2.1 引物的设计与合成 根据合成PPRV-N序列,自行设计引物并由宝生物工程(大连)有限公司合成。为了便于构建表达载体,在上下游的5′端分别加入HindⅢ、NdeⅠ酶切位点,并在下游3′端加入终止密码子TAA(表1)。
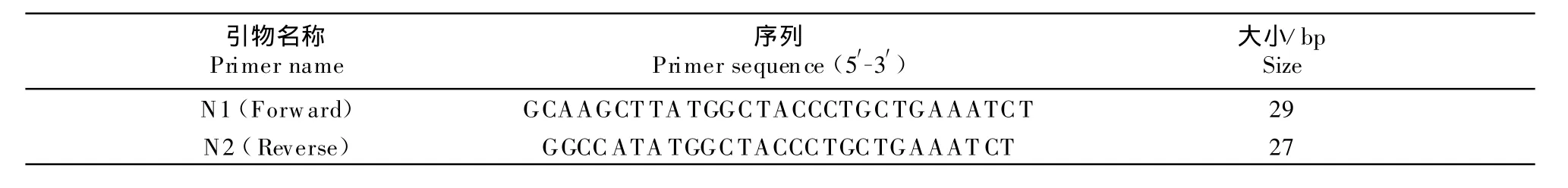
表1 引物设计Table 1 primer design
1.2.2 目的片段的扩增及表达载体的构建 以公司合成质粒为模板,N1、N2为引物,PCR扩增以获得目的基因。反应体系25 μ L:上游引物N1 0.5 μ L、下游引物N2 0.5 μ L、模板DNA 1 μ L、10×PCR buffer 2 μ L、dNTP 2 μ L、Tag酶0.25 μ L、ddH2O 19 μ L;反应条件:95℃4 min;94℃30 s,58℃40 s,72℃2 min,35个循环;72℃延伸10 min。在10 g/L的琼脂糖凝胶中电泳后,分离回收扩增产物。将回收产物与pMD18-Ts在4℃连接过夜,将连接产物转化至JM109感受态细胞,转化产物均匀涂布于含有氨苄青霉素(100 mg/mL)的LB琼脂平板上,37℃培养过夜,挑取白色单菌落増菌培养后提取质粒。对提取的质粒进行PCR和酶切鉴定,将鉴定为阳性的重组菌的质粒送大连华大生物技术有限公司进行测序。将测序正确的克隆命名为pMD-18Ts-N。将pMD-18Ts-N与pET-28a(+)用HindⅢ、NdeⅠ进行双酶切,回收目的片段,按照目的片段5 μ L、载体片段3 μ L、连接buffer 2 μ L、T4连接酶0.5 μ L的体系4℃连接过夜,取连接产物转化JM109感受态细胞,在含有卡那霉素(50 mg/mL)的LB琼脂平板上,37℃培养过夜,挑取单菌落在LB(含50 mg/mL卡那霉素)中振荡培养,碱裂解法提取质粒后,进行PCR与双酶切鉴定,两者均为阳性时将所构建的重组表达质粒命名为pET28a-N。
1.2.3 重组蛋白表达菌株的确定 将筛选出的阳性重组质粒转化原核表达菌Transetta(DE3)中,转化产物均匀涂布于含有卡那霉素(100 mg/mL)的LB琼脂平板上,37℃培养过夜,挑取白色单菌落増菌培养后提取质粒。对提取的质粒进行PCR和酶切鉴定,将鉴定为阳性的重组菌的质粒送大连华大生物技术有限公司进行测序。将测序正确的菌株命名为Transetta-pET28a-N。
1.2.4 重组蛋白表达 筛选出的阳性菌株Transetta-pET28a-N,涂布于含有卡那霉素抗性的LB培养基37℃过夜培养,次日挑取菌落于含有卡那霉素抗性液体LB培养基中37℃、200 r/min振荡培养至对数中期(A600=0.5~1.0)时加入终浓度为1.0 mmol/L的IPTG,诱导6 h后,离心收集菌液,用SDS-PAGE电泳分析斑白表达情况。
1.2.5 重组蛋白表达诱导条件的确定 将鉴定表达的菌株按照1%的比例接种于含有卡那霉素抗性的LB液体培养基中,37℃、200 r/min振荡培养至对数中期(A600=0.5~1.0)时,在诱导温度和时间不变的情况下改变诱导剂的浓度,分别为0.1、0.5、1、2 mmol/L进行诱导表达,再在浓度不变的情况下改变诱导的时间分别为4、5、6 h,最后在诱导剂浓度和诱导时间不变的情况下改变诱导的温度,分别为28、30、37℃,收集细菌,进行SDS-PAGE分析对比后确定最终诱导条件为28℃、IPTG浓度为1 mmol/L、200 r/min、6 h。
1.2.6 表达产物的鉴定 首先鉴定融合蛋白上的HIS标签蛋白,收集最佳诱导条件诱导的菌液进行SDS-PAGE电泳,将表达产物电转移至硝酸纤维素膜上作为抗原,用含50 g/L的脱脂奶粉的培养液37℃封闭1 h,用TBST洗涤3次,每次10 min,1∶1 000稀释鼠抗His标签单抗为一抗,37℃作用1 h,用TBST洗涤3次,每次10 min,再用1∶2 000稀释辣根过氧化物酶标记的羊抗鼠IgG为二抗,37℃作用1 h,用TBST洗涤3次,每次10 min,最后用DAB显色液显色。再用羊抗PPRV Nigeria75/1株多克隆抗体为一抗,辣根过氧化物酶标记的兔抗山羊IgG为二抗进行Western blot,对表达的蛋白进行免疫活性分析。
1.2.7 表达产物的可溶性分析 在扩大表达菌液量之后,取500mL表达菌液进行离心(10 000 r/min,2 min)回收沉淀,用重悬液按照1∶25倍的比例重悬。进行超声,超声后的上清与沉淀分别回收,各取40 μ L进行SDS-PAGE与Western blot,对比超声前、超声后上清、超声后沉淀中融合蛋白的量。
2 结果
2.1 N基因的扩增
用设计的引物对合成的质粒进行PCR扩增,扩增引物用10 g/L琼脂糖凝胶电泳检测,扩增出一条1 500 bp左右的条带(图1),与预期结果一致。
2.2 N基因重组表达载体的构建与鉴定
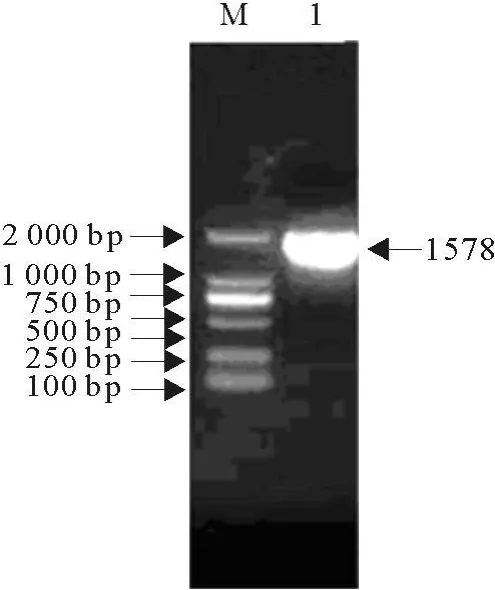
图1 PPRV N基因的PCR鉴定Fig.1 PCR identification of PPRV N gene
回收PCR产物,与pMD-18t载体连接,对阳性菌落扩大培养后小型抽提质粒并用HindⅢ、NdeⅠ双酶切鉴定(图2A)及送华大基因公司测序,确定扩增的N基因的正确性。对酶切鉴定及测序正确的质粒与pET-28a载体定向重组,并对重组后的质粒进行双酶切鉴定(图2B)及送华大基因公司测序,以确定N基因序列的完整。

图2 重组质粒的酶切鉴定 Fig.2 Identification of the recombinant plasmid by digestion with enzymes
2.3 N蛋白的表达与鉴定
2.3.1 蛋白表达诱导条件的确定 经过对诱导温度,时间及诱导剂IPTG的用量的筛选,确定最终反应条件为28℃、IPTG浓度为1 mmol/L、200 r/min、6 h(图3)。
2.3.2 HIS标签单抗进行Western bolt检测 表达产物经SDS-PAGE电泳后转移至NC膜上进行Western blot检测,结果显示,出现3条特异性条带,而对照组未出现条带(图4)证明重组融合蛋白上含有标签蛋白,利于以后的蛋白纯化。
2.3.3 融合蛋白活性的鉴定 表达产物经SDSPAGE电泳后转移至NC膜上进行Western blot检测,结果显示特异性条带与HIS标签Western blot检测结果相同,表明表达的蛋白能被PPRV阳性血清所识别,具有与阳性血清反应的活性(图5)。
2.3.4 表达产物的超声裂解与分析 在扩大表达菌液量之后,取500 mL表达菌液进行离心(10 000 r/min、2 min)回收沉淀,用重悬液按照1∶25倍的比例重悬。进行超声,超声后的上清与沉淀分别回收,各取40 μ L进行SDS-PAGE与Western blot,对比超声前、超声后沉淀上清、超声后中融合蛋白的表达量及活性。结果表明目的蛋白为可溶性蛋白(图6)。
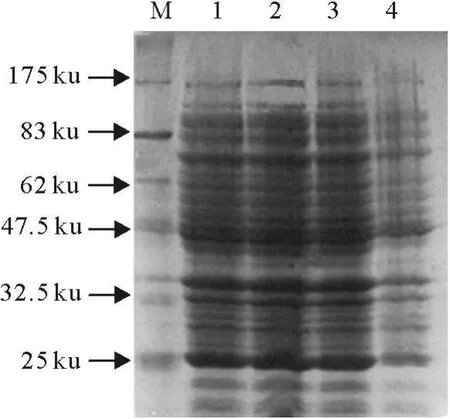
图3 SDS-PAGE分析pET28a-N的表达Fig.3 Ex pression of pET28a-N analyzed by SDS-PAGE

图4 表达产物的免疫印迹分析Fig.4 Analysis of the expressed fusion protein by Western blot

图5 表达产物的免疫印迹分析Fig.5 Analysis of the ex pressed fusion protein by Western blot
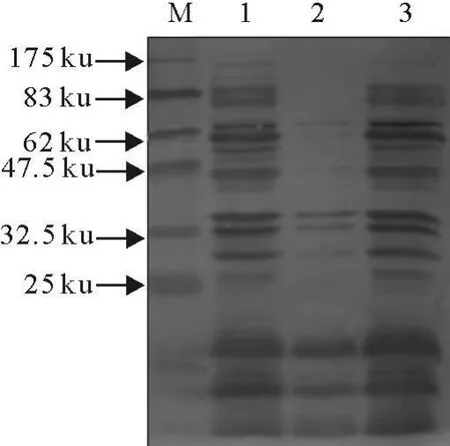
图6 表达产物超声裂解后的分析 Fig.6 Analysis of the expressed product with sonication
3 讨论
目前,国际上针对小反刍兽疫的诊断技术主要有竞争ELISA[7]、夹心ELISA[8]、RT-PCR[9]等。我国现在对小反刍兽疫的研究还仍处于起步阶段,所以特异性高、使用简便、成本低的检测手段的研发就显得尤为重要。N蛋白的抗原稳定性高,而基因的保守与特异性是建立ELISA方法的关键。本试验对GenBank发表的PPRV Nigeria 75/1株N基因序列进行优化、克隆及原核表达,旨在为后期建立间接ELISA的快速检测手段提供物质基础。
原核表达系统具有操作简便、表达量高的显著优点,但表达产物大多以无生物学活性、不溶性的包涵体形式存在。大多研究表明,多数外源蛋白在大肠埃希菌内不能正确折叠获得天然空间结构和生物学功能,以无活性的形式存在。需先将包涵体溶解后进行重组蛋白复性,这些过程操作复杂,极大增加了获得高活性纯品的难度和提纯成本。
本试验选用的是高效表达载体pET-28a(+),pET-28a(+)中的HIS标签蛋白在纯化过程中起到很重要的作用,为今后的蛋白纯化提供保障。在诱导表达的过程中,通过对诱导剂的浓度、诱导温度诱导时间的调节,最终确定诱导表达的最佳条件为28℃、IPTG的浓度为1 mmol/L、200 r/min、诱导6 h获得了可溶性表达的PPRV N抗原蛋白,再进行Western blot检测目的蛋白能与羊抗PPRV Nigeria75/1株多克隆抗体结合,并出现特异性条带,说明表达的重组蛋白具有免疫活性,为建立实验室快速诊断技术奠定了基础,也为新型基因工程疫苗[10]的研制提供了依据。
[1] Forsyth M A,Barrett T.Evaluation of polymerase chain reaction for the detection and charactefisation of finder pest and peste des petits ruminants viruses for epidemiological studies[J].Virus Res,1995,39(2-3):151-163.
[2] 李井春,赵凤菊,赵玉军,等.小反刍兽疫概况[J].黑龙江畜牧兽医,2004,12(2):63-64.
[3] Ismail T M,Yamanaka M K,Saliki J T,et al.Cloning and expression of the nucleoprotein of peste des petits ruminants virus in bacteria for use in serological diagnosis[J].Virology,1995,208(2):776-778.
[4] Libeau G,DialloA,Colas F,et al.Rapid differential diagnosis of rinder-pest and peste des petits ruminants using an immunocapture ELISA[J].Vet Res,1994,134(12):300-304.
[5] Libeau G,Prehaud C,Lancelot R,et al.Development of a competitive ELISA for detecting antibodies to the peste des petits ruminants virus using a recombinant nucleoprotein research[J].Vet Sci,1995,58(1):50-55.
[6] 王志亮,包静月,吴晓东,等.我国首例小反刍兽疫诊断报告[J].中国动物检疫,2007,24(8):24-26.
[7] Sreenivasa B P,Singh R P,Mondal B,et al.Marmoset B95a cells:a sensitive system for bacteria of peste des petits ruminants virus[J].Vet Res Comm,2006(30):103-108.
[8] Singh R P,Sreenivasa B P,Dhar P,et al.Development of a monoclonal antibody based competitive-ELISA for detection and titration of antibodies to peste des petits ruminants virus[J].Vet Microbiol,2004,98(1);3-15.
[9] Balamurugan V,Sen A,Saravanan P,et al.One-step multiplex RT-PCR assay for the detection of peste des petits ruminants virus in clinical samples[J].Vet Res Comm,2006,30(6):655-666.
[10] Bailey D,Banyard A,Dash P,et al.Full genome sequence of peste des petits ruminants virus,a member of the Morbillivirus genus[J].Virus Res,2005,110(1-2):119-120.
猜你喜欢
特产研究(2024年1期)2024-03-12 05:40:56
天然产物研究与开发(2019年10期)2019-11-05 10:12:44
食品科学(2018年10期)2018-05-23 01:27:28
河南畜牧兽医(2017年20期)2018-01-19 02:48:17
西南医科大学学报(2015年1期)2015-08-22 13:01:46
河南畜牧兽医(2015年13期)2015-08-15 00:46:58
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
当代畜禽养殖业(2014年6期)2014-02-27 07:59:09
当代畜禽养殖业(2014年6期)2014-02-27 07:59:06
