
应用PCR方法向基因内部引入序列的策略研究
2011-03-07 04:55:40徐亚英
东北农业大学学报 2011年4期
徐亚英,王 勇,罗 宏,陈 典
(东北农业大学园艺学院,哈尔滨 150030)
基因定位突变是指将某一克隆基因按预先设计好的目标进行定点改造,它仅适用于基因序列已知,蛋白结构与功能的关系比较明确的基因[1]。由于它具有简单易行、重复性高等优点,现已发展成为基因操作的一种基本技术[2]。常见体外定点突变的方法有化学诱变、寡核苷酸介导的诱变、盒式诱变和基于PCR方法的诱变等[3],其中基于PCR方法的诱变以其简单、快速的优势逐渐成为DNA体外修饰产生新型蛋白质的最常用方法[4]。
Cre重组酶来源于P1噬菌体[5],它不仅具有催化活性,而且同限制酶相似,可以识别特异的DNA序列并进行特定的剪切和拼接[6-7]。当其作用序列位点—两个34bp的loxP位点存在时,它可根据loxP位点的方向不同发生倒位、移位和删除反应[8-9]。Cre/1oxP已广泛应用于内、外源基因失活或敲除、基因激活等反应[10-11]。目前,Cre/1oxP定位重组系统在应用中存在两个主要问题:Cre重组酶的删除效率较低,其删除效率仅为50%左右[12];此前,在我们实验室的研究工作中发现,当Cre重组酶与loxP位点共存于一个寄主细胞中时,常由于Cre重组酶的痕量表达而引起loxP位点间序列的非目的性删除。针对以上缺陷,本试验设计了3条引物,利用PCR方法向cre基因内部引入一段具有增强基因在真核生物中表达效率的内含子序列(pfu1305.1)[13],以改变其在原核细胞中的通读序列,完成cre基因的改造,进而达到提高Cre/1oxP定位重组系统在转基因真核生物中的删除效率的目的同时,也避免了cre基因的渗露表达。为了使cre基因的突变达到最佳效果,本试验设计了三种PCR策略,并对每种策略进行了体系优化,以确定基因改造的最佳方法。
1 材料与方法
1.1 材料
1.1.1 质粒
pMM23、pCAMBIA1305.1均为本实验室保存,其中pMM23含有Cre序列(1029bp),pCAMBIA1305.1含有需插入到Cre中的内含子序列(AF354045.1,190bp)。
1.1.2 试剂
Pfu DNA聚合酶(附带Buffer及Mg2+)、dNTPs(购自上海生工);DNAmarker(DL2000,购自宝泰克)。
1.1.3 PCR引物序列
依据Cre(X03453.1)和intron(AF354045.1)的序列,设计了如下三条引物用于cre基因的改造,所有引物均由上海生工合成(见表1)。配成100 mmol·L-1的储存液并稀释成10 mmol·L-1备用。
1.2 方法
利用三种PCR策略对cre基因进行突变的反应体系、条件及检测。
1.2.1 一步PCR策略
将三条引物08crein1、crein2、cre3与两个模板pCAMBIA1305.1、pMM23加入同一反应体系中,20μL扩增体系中包括:10×PCR buffer 2.0μL;dNTPs(10 mmol·L-1)0.4μL;08crein1(10 mmol·L-1),cre3(10 mmol·L-1) 各 1μL; pMM23(20 ng·μL-1)0.6μL;然后分别对Mg2+、中间引物crein2、内含子扩增模板pCAMBIA1305.1、Pfu进行了不同浓度的单因素体系优化试验;Mg2+(25 mmol·L-1)浓度设置为1.0、1.5、2.0、2.5、3.0μL;crein2(1 mmol·L-1),0.1、0.2、0.5、0.8、1μL;pCAMBIA1305.1 (20 ng·μL-1),0.06、0.12、0.5μL;Pfu(2.5 U·μL-1),0.2、0.4、0.6μL;加ddH2O补足20μL。当就某一成分进行体系优化时,其他成分取中间值。扩增条件:94℃,7 min;30个循环(94℃,30 s;52℃,40 s;72℃,2 min 30 s);72℃,7 min。
扩增产物用1%的琼脂糖凝胶电泳检测。电泳缓冲液为1×TAE,保持100 V恒压(4~5 V·cm-1),电泳30 min,紫外灯下检测扩增结果。
表1 cre基因改造的引物Table1 Primers of cre gene transformation
1.2.2 二步PCR策略
利用PCR方法分两步对cre基因进行突变。
①Intron的扩增
20μL扩增体系中包括:10×PCR buffer,2.0μL; MgCl2(25 mmol·L-1),2.0μL; dNTPs(10 mmol·L-1),0.2μL; 08crein1(10 mmol·L-1)、crein2(10 mmol·L-1)各 1μL; pCAMBIA1305.1(100 ng·μL-1),0.2μL;Pfu(2.5 U·μL-1),0.2μL;加ddH2O补足20μL。
扩增条件:94℃,7 min;30个循环(94℃,30 s;56℃,30 s;72℃,30 s);72℃,7 min。
电泳分析操作同上,扩增得到的目的片段约为220bp(内含子加两端引物的长度),命名为Pfu1305.1,此步PCR产物作为下一步反应引物。
②crein的扩增:以pMM23为模板,以Pfu 1305.1、08crein1、cre3为引物进行扩增反应。
20μL 扩增体系中包括:10×PCR buffer 2.0μL; dNTPs(10 mmol·L-1)0.2μL; 08crein1(10 mmol·L-1);cre3(10 mmol·L-1)各 1μL;pMM23(20 ng·μL-1)0.2μL;Pfu(2.5 U·μL-1)0.2μL;然后分别对Mg2+、单引物Pfu1305.1进行了不同浓度的单因素体系优化试验;Mg2+(25 mmol·L-1)浓度设置为0.5、1.0、1.5、2.0、2.5μL;Pfu1305.1(20 ng·μL-1),0.2、0.5、0.8μL;加ddH2O补足20μL。当就某一成分进行体系优化时,其他成分取中间值。
扩增条件:94℃,7 min;30个循环(94℃,30 s;52℃,40 s;72℃,2 min 30 s);72℃,7 min。电泳分析操作同上。
1.2.3 三步PCR策略:
①同1.2.2中的①;
②以pMM23为模板,以Pfu1305.1为引物对cre基因进行单引物扩增;
20μL 扩增体系中包括:10×PCR buffer,2.0μL; MgCl2(25 mmol·L-1),2.0μL; dNTPs(10 mmol·L-1),0.2μL; Pfu1305.1(20 ng·μL-1),0.5μL;pMM23(20 ng·μL-1),0.2μL;Pfu(2.5 U·μL-1),0.2μL;加ddH2O补足20μL。
扩增条件如下:94℃,7 min;5 or 10 or 15 or 20(94℃,30 s;52℃,40 s;72℃,2 min 30 s)
③取1μL上述扩增产物(crein基因)为模板,以08crein1、cre3为引物对插入内含子序列的cre基因进行PCR扩增。
20μL 扩增体系成分如下:10×PCR buffer,2.0μL;MgCl2(25 mmol·L-1),2.0μL;dNTPs(10 mmol·L-1),0.2μL;08crein1(10 mmol·L-1)、cre3(10 mmol·L-1)各1μL;crein,0.2μL; Pfu(2.5 U·μL-1),0.2μL;加ddH2O补足。
扩增条件:94℃,7 min;30个循环(94℃,20 s;56℃,30 s;72℃,1 min);72℃,7 min。电泳分析操作同上。
2 结果与分析
2.1 一步PCR策略
将三条引物08crein1、crein2、cre3与两个模板pCAMBIA1305.1、pMM23加入同一反应体系中进行基因扩增,结果见图1。

图1 一步PCR策略基因扩增结果Fig.1 Amplification result of the gene by one-step PCR
不同模板比例、不同酶加入量、不同中间引物浓度、不同Mg2+浓度下的部分扩增结果见图1。如前文所述,改造后的crein基因大小应为1200bp左右,但图中显示,对不同因素进行优化,在1200bp处都无可见条带,而在270bp处有较暗杂带产生,说明利用三引物两模板的一步PCR策略,即便是对PCR反应体系进行优化,也难以扩增出目的条带。
2.2 两步PCR策略
2.2.1 内含子的扩增
以pCAMBIA1305.1为模板、08crein1和crein2为引物进行内含子的扩增结果如图2所示,在220bp处有较亮的目的条带,且不存在非特异扩增。此步特异扩增效率高,故不必对体系进行优化。
2.2.2 crein的扩增
此步反应是以pMM23为模板,以Pfu1305.1、08crein1、cre3为引物进行目的条带的扩增。如图3所示,不同的Mg2+加入量和不同的Pfu1305.1浓度条件下均在1200bp处扩增出了较亮的目的条带,也均在约270与2500bp处有较暗的杂带产生,且不同体系条件下扩增结果基本一致。结果表明,应用两步PCR策略可以完成基因的改造,但存在非特异扩增(此步2500bp处的非特异条带不明显)。
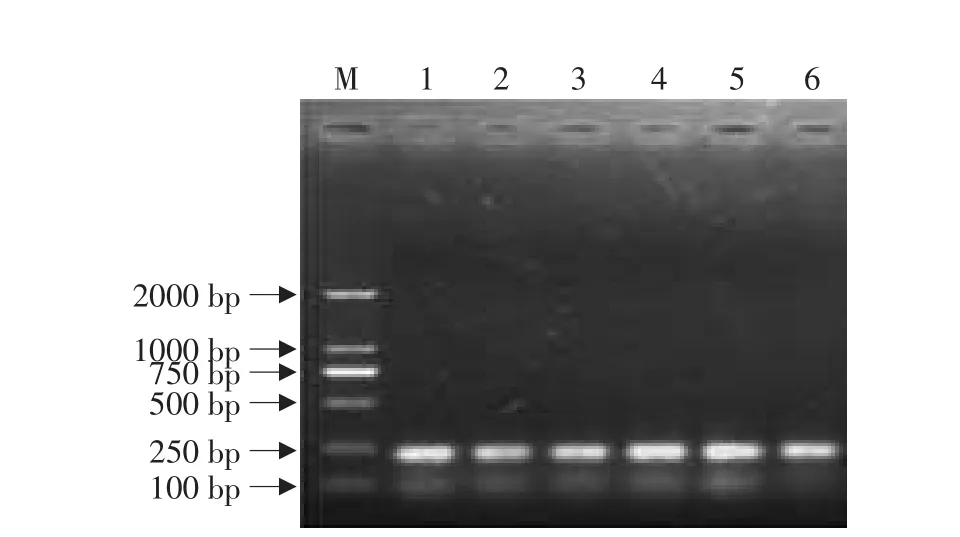
图2 Intron的扩增结果Fig.2 Amplification results of the intron
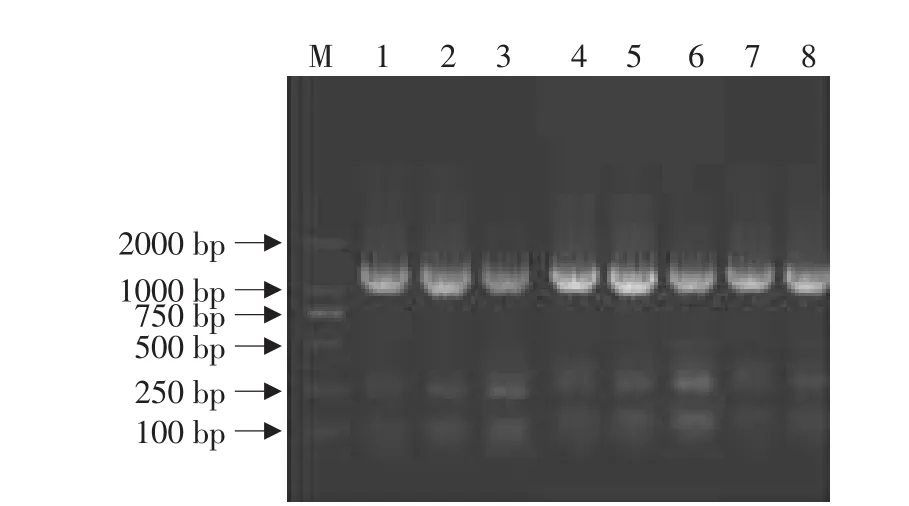
图3 不同条件下crein基因的扩增结果Fig.3 Amplification results of crein gene under different conditions
2.3 三步PCR策略
2.3.1 内含子的扩增
结果同2.2.1。
2.3.2 单引物扩增
此步骤是以pMM23为模板,以Pfu1305.1为引物对cre基因进行单引物扩增,以增大crein基因数量,并将其作为下步PCR反应的模板。在试验中,对PCR扩增的循环数进行了梯度设置,即分别为5、10、15、20个循环。设置循环的目的在于探讨单引物扩增循环数的不同是否会对下步crein基因序列的扩增产生影响。由于此步为单引物循环,循环数又较少,故凝胶电泳未显示出目的条带,如图4所示。
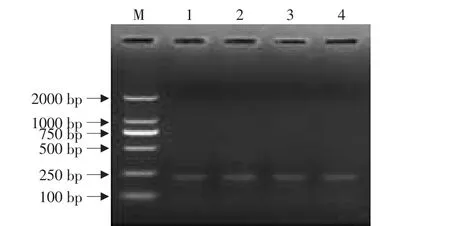
图4 crein基因的单引物扩增Fig.4 Amplification results of crein gene using a single primer
2.3.3 crein基因的扩增
结果见图5。

图5 crein基因的三步扩增结果Fig.5 Amplification results of crein gene using three-step PCR
分别各取上述不同循环的PCR产物1μL为模板,以08crein1、cre3为引物进行PCR扩增。结果如图5所示,当分别采用5、10、15、20个循环数进行模板扩增后,在酶用量分别为0.2和0.4μL条件下,都扩增出了明亮而特异的1200bp左右的目的条带。说明,单引物扩增后产生的微量产物可以作为本步扩增的模板,扩增结果并不因循环数的不同而产生明显差异。当DNA聚合酶的用量分别为0.2、0.4μL时,扩增条带的亮度也没有明显差异。为了减少前一步PCR过程对此步骤的影响,前一步单引物扩增循环数可取最小值,即5。
3 讨论
3.1 不同PCR策略的优劣分析
基因突变技术是蛋白质工程中应用的重要技术之一,包括单个碱基或整段序列的替换、基因片段的插入与删除等类型,可以有目的改变DNA序列中的碱基,使之满足不同的研究和应用需求[14]。近几年来,以PCR为基础的定点诱变方法得到不断的创新和改进,使得定点诱变越来越省时省力,简单易行,倍受研究者青睐[15]。而在本试验中,针对三种PCR策略进行比较分析,以确定基因改造的最佳方法,其优劣分析如下:
一步PCR策略的优点是整个基因改造过程只需一步即可完成,方便快捷,但在其反应体系中成分复杂,共有两个模板与三个引物,几个PCR反应同时进行,且相互间互相干扰,虽然我们就反应各组成体系及反应条件都进行了摸索与优化,但都没有得到理想结果。因此,一步PCR策略对反应条件和体系的要求较为苛刻,目的基因的扩增具有一定的难度;相对于一步PCR策略,二步PCR策略是先独立扩增出了用于基因突变的引物,然后再将其加入体系中,进行改造后基因的扩增。此种策略减少了一定量的基因扩增的干扰因素,但把单引物扩增和双引物扩增放在一个体系中进行,反应过程仍存在其他的干扰,因此,虽能扩增出较清晰的目的条带,但存在着较多的非目的条带扩增;而三步PCR策略分解了整个基因改造的全过程,相当于把每一步反应都独立出来,即将体系间的相互干扰降到最小。虽然其扩增步骤为三步,相对较为繁琐,但由于每一步反应都是比较简单,基本上不必进行任何体系的摸索与优化,就可以直接获得特异、明晰的目的条带,且反应条件较为宽泛,易于进行目的基因的扩增。因此,可认为三步PCR策略是三种基因改造策略中最实用的策略。其基因改造的路线如图6所示。

图6 利用三步PCR策略进行基因改造的路线Fig.6 Schematic diagram of three-step PCR approach for improving crein gene
3.2 三步PCR策略的引申
本试验针对在基因5′端插入序列的情况进行了探讨。当拟在基因中间部位插入序列时,可利用4个引物5步PCR的方法进行。第一步,双引物扩增得到基因前半段;第二步,单引物扩增获得含有插入序列的前半段基因;第三步,双引物扩增以产生基因后半段的部分碱基;第四步,单引物扩增得到目的基因;第五步,双引物扩增进行目的基因的累积。
4 结论
本研究以PCR方法在基因起始部位附近进行基因的突变或改造,最佳方法为三步PCR策略:首先是引物1与引物2引导的插入序列的扩增;其次是以扩增后所得序列为单引物进行的扩增,此步骤所得的产物可作为下步反应的模板;最后是以第二次PCR产物作为模板,以引物1和引物3所进行的改造后基因的指数扩增。
[1]赵和.生物技术在基因诱变中的应用[J].河北农业科学,2006,10(1):92-96.
[2]王秋颖.基因体外定点诱变技术[J].山西农业大学学报,2007,27(6):124-126.
[3]梁晖,李雪,孙紫阳.发生点突变的pCDNA3.1(+)质粒的构建[J].天津医科大学学报,2008,14(3):374-382.
[4]李晶琴.基于PCR技术的蛋白质改造策略[J].国际检验医学杂志,2006,27(12):1129-1131.
[5]Sternberg N,Sauer B,Hoess R,et al.Bacteriophage P1 cre gene and its regulatory region evidence for multiple promoters and for regulation by DNA methylation[J].Journal of Molecular Biology,1986,187:197-212.
[6]邱淑萍,陈在杰,王锋.Cre/loxp位点特异性重组系统在转基因植物中的应用[J].福建农业学报,2008,23(2):211-217.
[7]李文旭.基于Cre lox P系统诱导删除抗生素基因植物表达载体的构建[D].福州:福建农林大学,2009.
[8]李大旭,马静云,曹永长.Cre lox P重组系统的特点及其在抗体研究中的应用[J].生物技术通报,2008(1):56-57.
[9]王文棋,盖颖,陆海,等.DNA重组酶Cre介导载体间基因的重组转移[J].生物化学与生物物理进展,2007,34(11):1210-1215.
[10]任荣荣,王英伟.Cre/lox p系统的应用及进展[J].广州医学院学报,2008,36(2):78-80.
[11]陈双喜,许守明.Cre/lox P系统介导的位点特异性重组技术[J].安徽农学通报,2009,15(15):27-28.
[12]Wang Y,Chen B J,Hu Y L,et al.Inducible excision of selectable marker gene from transgenic plants by the Cre/lox site-specific recombination system[J].Transgenic Research,2005,14:605-613.
[13]丁红梅,邵根宝,徐银学.内含子与基因表达调控[J].畜牧与兽医,2006,38(3):50-52.
[14]徐芳,姚泉洪,熊爱生,等.重叠延伸PCR技术及其在基因工程上的应用[J].分子植物育种,2006,4(5):747-750.
[15]黄春晓,段学军.以PCR为基础的定点诱变方法研究进展[J].河南农业科学,2006(12):5-8.
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04 11:40:51
生物学通报(2022年1期)2022-11-22 08:12:18
南京林业大学学报(自然科学版)(2021年5期)2021-10-13 02:06:16
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01 08:10:30
生物工程学报(2019年6期)2019-07-10 08:38:38
生物学通报(2019年1期)2019-02-15 16:33:43
生物学通报(2018年12期)2018-10-10 06:52:36
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
广西林业科学(2016年3期)2016-03-16 05:43:25
中国光学(2015年1期)2015-06-06 18:30:20
