
LC/MS技术在寡核苷酸类药物生物样品定量分析和代谢物鉴定中的应用
2011-02-02 07:01钟大放陈笑艳
质谱学报 2011年1期
邓 泮,钟大放,陈笑艳
(中国科学院上海药物研究所,上海 201203)
LC/MS技术在寡核苷酸类药物生物样品定量分析和代谢物鉴定中的应用
邓 泮,钟大放,陈笑艳
(中国科学院上海药物研究所,上海 201203)
寡核苷酸是药物发展的新领域,这类药物是人工合成的DNA或RNA片段,一般由15~50个核苷酸组成,其药动学研究的经典方法是酶联免疫法,但由于该方法的建立耗时、耗资,因此成为新药研发的瓶颈。液相色谱-质谱(LC/MS)联用技术在寡核苷酸类药物生物分析中面临的主要问题有:待测物离子化效率低、基质效应严重、色谱分离条件与质谱不易兼容,样品处理回收率低,但该方法建立快速且选择性强,并能够同时进行代谢产物研究,因此,这项技术尽管存在一些缺点但依然备受关注。本文综述了过去15年中LC/MS技术在寡核苷酸类药物生物样品定量分析和代谢物鉴定中的发展和应用。
寡核苷酸药物;液相色谱-质谱联用;生物样品
在过去的10年中,寡核苷酸类药物已逐渐成为药物发展的新领域,这类化合物被证实可用于多种疾病的治疗,例如癌症、病毒细菌感染、自身免疫、感染性疾病、神经系统疾病以及代谢疾病[1-2]。该类药物是人工合成的DNA或 RNA片段,一般由15~50个核苷酸组成[3],其作用靶点为产生疾病蛋白质的基因。根据作用机理的不同可分为多类,其中,应用最广泛、开发前景最好的主要有两类:反义寡核苷酸(antisense oligonucleotides,AS-OGNs)和短干扰 RNA(si-RNAs)。AS-OGNs是 DNA或 RNA单链片段,其核苷酸序列可与靶mRNA或靶DNA杂交,抑制或封闭该基因的转录和表达,或诱导RNaseH识别并切割 mRNA使其丧失功能,从而发挥治疗作用[2]。si-RNAs是一种双链RNA,通过RNA干扰通路影响基因表达[4],与AS-OGNs相比,这类化合物的药用研究起步较晚,目前均处于研发阶段尚未有上市药物。已被美国FDA批准上市的AS-OGNs类药物有治疗年龄相关性黄斑变性的Macugen(美国 Eyetech公司,24 mer-OGN)和用于治疗巨细胞病毒引起视网膜炎的Fomivirsen(美国ISIS公司,21 mer-OGN),此外,尚有20余种 AS-OGNs类药物正处于临床研究阶段[5]。天然寡核苷酸进入体内后易被降解,因此只有通过化学修饰提高稳定性才能达到药用目的。已上市和处于开发阶段的寡核苷酸类药物主要是硫代磷酸寡核苷酸(phosphorothioate oligonucleotides,PS-OGNs)和2’-烷氧基修饰的混合骨架寡核苷酸(mixedbackbone oligonucleotides,MBOs)[6],典型核苷酸基团示于图1。在 PS-OGNs的磷酸二酯键中,1个非桥键的氧原子被硫原子所取代,经硫代修饰后的寡核苷酸可有效抵抗核酸酶的降解,提高药物的血浆蛋白结合率及其在组织中的分布,同时也会延缓药物的肾脏排泄[7]。MBOs是在核糖的2’位末端进行修饰,其中以2’-O-甲基和2’-O-甲氧乙基修饰最具代表性。MBOs具有更强的靶RNA亲和性和更好的代谢稳定性[8]。其他磷酸骨架和核糖修饰的寡核苷酸包括吗啉代的寡核苷酸、肽核酸和锁核酸等,目前这些寡核苷酸的应用有限[9]。
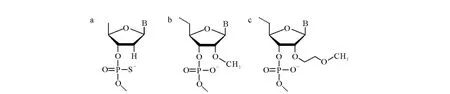
图1 结构改造后的典型核苷酸基团Fig.1 Representative modified nucleic acid building blocks
在临床前及临床研究阶段,定量分析生物样品中的寡核苷酸,以及鉴定代谢产物在其药代动力学(PK)、药动学/药效学(PK/PD)和毒代动力学(TK)评价中是至关重要的,这不仅为寡核苷酸药物提供PK、TK及代谢途径等重要信息,还对这类药物的结构改造起到指导作用,达到改善药物稳定性和降低清除率的目的。目前,寡核苷酸的生物样品定量分析方法包括基于杂交技术的酶联免疫法(ELISA)、放射性同位素法、毛细管凝胶电泳法(CGE-UV/荧光)和高效液相色谱法(HPLC-UV/荧光),这些方法在应用中都有其各自的特点及局限性[10]。其中,ELISA法以其高灵敏度和高通量的显著优势在寡核苷酸药物的 PK/TK研究中获得了最为广泛的应用[11-12],但它无法区分全长的寡核苷酸和核酸水解的代谢产物,易产生交叉杂交的现象,导致原药浓度被高估[10]。此外,为获得专属性试剂,ELISA方法的开发周期长(6~12个月)、成本高,通常成为寡核苷酸药物临床前和临床研究中的瓶颈。
液相色谱-质谱(LC/MS)联用技术将具有高分离能力的液相色谱法与高灵敏度和选择性的质谱法结合起来,虽然该技术已在小分子化合物和多肽的生物样品分析中得到广泛应用,但由于寡核苷酸类药物分子的多种特性(如酸性强极性、离子化效率低、易产生大量的金属加合离子),以及由这些性质导致的生物样品回收率不稳定和LC/MS分析中存在的严重基质效应、灵敏度低等问题,都成为LC/MS生物样品分析方法开发中需要攻克的难关[5,13]。本工作将对LC/MS技术在寡核苷酸类药物生物样品定量分析和代谢物鉴定中的应用进行综述。
1 寡核苷酸类药物生物分析中的LC/MS方法开发
1.1 质谱检测
1.1.1 离子化 目前,寡核苷酸类药物在临床上多采用静脉或皮下给药方式,给药后药物在体内迅速分布,然后进入消除相,消除相的血药浓度很低且维持时间较长,因此,为了准确描述消除相药时曲线,定量分析方法需要较高的灵敏度。ELISA法的灵敏度在众多定量分析方法中最高,可达1μg/L以下,能够检测到给药后170 h的血药浓度[12]。寡核苷酸属于酸性强极性化合物,结构中的每个磷酸二酯键上都带有1个酸性质子(p Ka~1)[14],通常这类化合物在 ESI源负离子模式下的响应较好,此外,由于这类化合物含有至少15个核苷酸基团,每个磷酸二酯键上的酸性质子在ESI软电离过程中均可能发生去质子反应,因此,在 ESI(-)检测条件下形成多重电荷形式的离子。根据这些特点,利用常规质荷比范围的质谱仪即可实现这类大分子离子的测定(>6 000 u),但这种多重质谱峰的存在使质谱信号被分散到了多个前体离子上,并且由于寡核苷酸的磷酸骨架带有多个阴离子,很容易结合Na+、K+等金属阳离子,进一步分散了质谱信号[14-15]。因此,对这类化合物质谱条件优化的目的,一方面是改善待测物的多电荷分布,针对丰度较高的前体离子优化质谱参数;另一方面是减少待测物在质谱分析时加合离子的形成。影响寡核苷酸离子化效率的因素主要包括溶液p H值,阳离子浓度和有机溶剂组成[5]。寡核苷酸是酸性化合物,使用质谱兼容的有机碱(如三乙胺(TEA))有助于提高质谱响应,但由于寡核苷酸能够与TEA通过离子键和作用形成“假中性分子”,造成寡核苷酸的电荷被屏蔽,使其在ESI源气化过程中难以通过库伦爆炸从小液滴中释放出来成为气态离子,从而产生信号抑制的现象[5]。Apffel等[16]于1997年发现,采用六氟异丙醇(HFIP)/TEA的缓冲液体系能够极大地提高寡核苷酸类化合物的质谱响应,自此之后,HFIP/TEA成为LC/MS分析寡核苷酸的常用缓冲液体系[17-20]。其原理是 HFIP(p Ka~9,bp=57℃)在p H 7.0的条件下呈分子状态自由挥发,在离子化和去溶剂过程中,液滴表面的HFIP挥发殆尽,p H值升高至10,在这样的条件下,寡核苷酸-TEA离子对能够顺利解离,促使寡核苷酸气态离子的生成[16]。此外,有机碱TEA还能够有效抑制金属加合离子的形成[21]。Apffel等[16]对比了 4种不同溶剂体系下,50 pmol寡聚胸腺嘧啶核苷酸dT20溶液的质谱响应 ,包括 400 mmol/L HFIP/TEA(p H 7),水(p H 7),100 mmol/L醋酸三乙胺(TEAA)水溶液和25 mmol/L TEA水溶液(p H 10),示于图2。结果显示,使用400 mmol/L HFIP/TEA溶液体系时,dT20的质谱响应是使用25 mmol/L TEA水溶液的2倍,使用 TEAA时质谱响应极低,而使用水作为溶剂时,检测到大量的加合离子。
由于寡核苷酸色谱分离中通常使用水相比例较高的流动相体系,因此柱后灌流有机溶剂(乙腈、甲醇或异丙醇)是改善这类化合物离子化效率、提高质谱响应的有效途径[5]。此外,柱后灌流0.1 mol/L咪唑乙腈溶液也能显著改善寡核苷酸的质谱响应[22],但其机理有待进一步研究。
1.1.2 质量分析器 在寡核苷酸的LC/MS分析中,常见的质量分析器是三重四极杆质谱、离子阱质谱和飞行时间质谱。三重四极杆质谱具有离子传输效率高、动态范围宽和灵敏度高的特点,在定量分析中具有显著的优势[18-19,23-25]。寡核苷酸在三重四极杆质谱中经过碰撞诱导解离主要产生嘧啶和磷酸的碎片离子,对序列分析没有帮助。一种24个核苷酸的硫代寡核苷酸(PF-ODN,5’-TCGTCGTTTTGTCGTTTTGTCGTT-3’)在 API 3000、API 4000、API 5000和Agilent 6460四种型号的三重四极杆串联质谱仪形成的碎片离子均比较单一,主要是m/z95磷酸离子和m/z125胸腺嘧啶碎片离子,示于图3[18-19]。另一种18个核苷酸的2’-烷氧基取代的硫代寡核苷酸在API 4000质谱仪上得到的主要碎片离子质荷比同样是m/z95和125[17],且产生这两种碎片离子所需的碰撞能量均较高。
图2 寡聚胸腺嘧啶核苷酸dT20溶液(50 pmol)在不同溶剂中ESI总离子流响应对比[16]Fig.2 Comparison of ESI total ion currents of 50 pmol dT20 in different solvents[16]
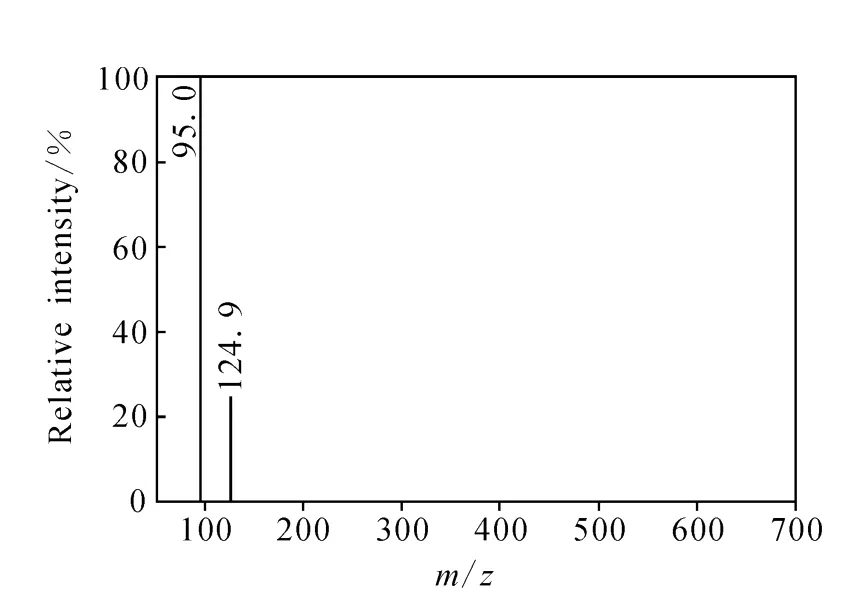
图3 硫代寡核苷酸PF-ODN在三重四极杆串联质谱分析时的碎片离子质谱图[19]Fig.3 Mass fragments of PS-ODN on triple quandrople mass spectrometry[19]
离子阱质谱能够产生与寡核苷酸序列相关的w和a-B系列的诊断性离子[26],以硫代寡核苷酸为例,其在离子阱质谱上的裂解方式示于图4。其中,w系列离子的末端是5’-磷酸,用来判断3’→5’方向的核苷酸序列;a-B系列离子末端是3’-呋喃,用来判断 5’→3’方向的核苷酸序列[20,27-28],这些离子不仅可以用来对寡核苷酸进行定量,还可以用于鉴定其代谢产物[20]。Dai等[20]使用离子阱质谱得到一种18个核苷酸的硫代寡核苷酸(G3139,5’-TCTCCCAGCGTGCGCCAT-3’)的w和a-B系列碎片离子,并将其作为产物离子用于定量分析。Simple Oligonucleotide Sequencer(SOS)软件能够通过w和a-B的质谱碎片离子快速分析核酸序列[29],目前已有关于此软件在代谢物鉴定中应用的报道[27]。L TQ Orbitrap质谱能够通过高分辨数据确认代谢物核苷酸的组成,区分质量相差不到1 u的代谢物[30],但这类离子阱质谱检测器的灵敏度和动态范围均不及四极杆质谱。飞行时间质谱主要用于寡核苷酸的定性研究,目前未见其在定量分析中应用的报道。
不论使用何种检测器,TEA已经成为寡核苷酸质谱分析中必不可少的添加剂,但 TEA属于难挥发性碱,易吸附在质谱检测器上,造成仪器污染并导致质谱检测灵敏度显著下降,这成为质谱仪在寡核苷酸类药物生物分析应用中的限制因素。为保证质谱仪的正常运作,使用 TEA对寡核苷酸进行质谱分析时,需要及时清洗质谱检测器,或在不影响检测灵敏度的前提下适当提高气帘气的流量,尽量减少进入质谱仪的 TEA浓度[24]。
1.2 与质谱兼容的反相离子对色谱法
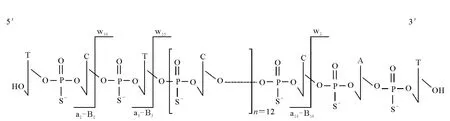
图4 硫代寡核苷酸在离子阱质谱分析时的质谱断裂方式Fig.4 Mass fragment pattern of phosphorothioate oligonucleotide on ion-trap mass spectrometry
寡核苷酸为酸性强极性化合物,在普通反相色谱柱上几乎无保留,为增加这类化合物的色谱保留,可以在流动相中加入适当的碱性离子对试剂与寡核苷酸离子结合形成离子对,此离子对在流动相中不易解离,而在反相C18键合相中保留,进而在固定相和流动相间进行分配,最终实现色谱分离[14,31]。这种反相离子对色谱法(IPRP-HPLC)已成为寡核苷酸色谱分析的首选技术。早期的IP-RP-HPLC-UV分析方法使用醋酸三乙胺作为流动相中的离子对试剂,带正电的三乙胺与寡核苷酸阴离子形成离子对,显著改善了寡核苷酸在反相色谱柱上的保留和峰形[32]。流动相体系中醋酸三乙胺的浓度通常为50~100 mmol/L(p H 7.0)。在醋酸三乙胺的缓冲液体系中,醋酸的p Ka 4.75,沸点较高为118℃,在p H 7.0的条件下几乎完全处于解离状态,因此在质谱离子源的去溶剂过程中无法挥发,使寡核苷酸难以从小液滴中释放出来成为气态离子,产生明显的信号抑制现象[16]。与醋酸三乙胺性质类似的重碳酸三乙胺(TEAB)也被用于IP-RP-HPLC法分离寡核苷酸,但与醋酸三乙胺存在同样的质谱信号抑制现象[33]。这两种离子对试剂虽然已经普遍应用于寡核苷酸的 HPLCUV分析中[32,34-35],但由于其与质谱的兼容性不佳,在寡核苷酸的LC/MS定量分析中均未能获得广泛的使用。此外,醋酸三乙胺对寡核苷酸混合物的分离能力也不及 TEA/HFIP流动相体系[36]。
TEA/HFIP成为LC/MS法分析寡核苷酸时的首选离子对试剂缓冲液体系。在色谱分离中,TEA发挥着离子对试剂的作用,其浓度决定寡核苷酸的色谱保留行为[31,37]。Gilar等[32]研究了不同的 TEA缓冲液体系对碱基数为10~30的寡核苷酸混合物的分离能力,示于图5。结果显示,使用含有16.3 mmol/L TEA/400 mmol/L HFIP(p H 7.9)的缓冲液体系分离效果最好,其次为 4.1 mmol/L TEA/100 mmol/L HFIP(p H 8.2)体系,而在含有 0.1 mol/L TEAA(p H 7.0)的流动相中存在分离度不佳的寡核苷酸共流现象。在质谱检测中,TEA从两方面提高寡核苷酸的质谱响应:1)作为有机碱提高酸性寡核苷酸的去质子效率;2)抑制寡核苷酸金属加合离子的产生。若流动相中离子对试剂浓度低,可能导致寡核苷酸保留效果不佳、分离度差或不能有效去除质谱检测中金属加合离子的干扰;若浓度过高,可能直接抑制质谱响应,因此在优化流动相中水相的离子对试剂和有机溶剂时,需要综合考虑色谱分离和质谱响应两方面因素,并最终在二者之间达成平衡。
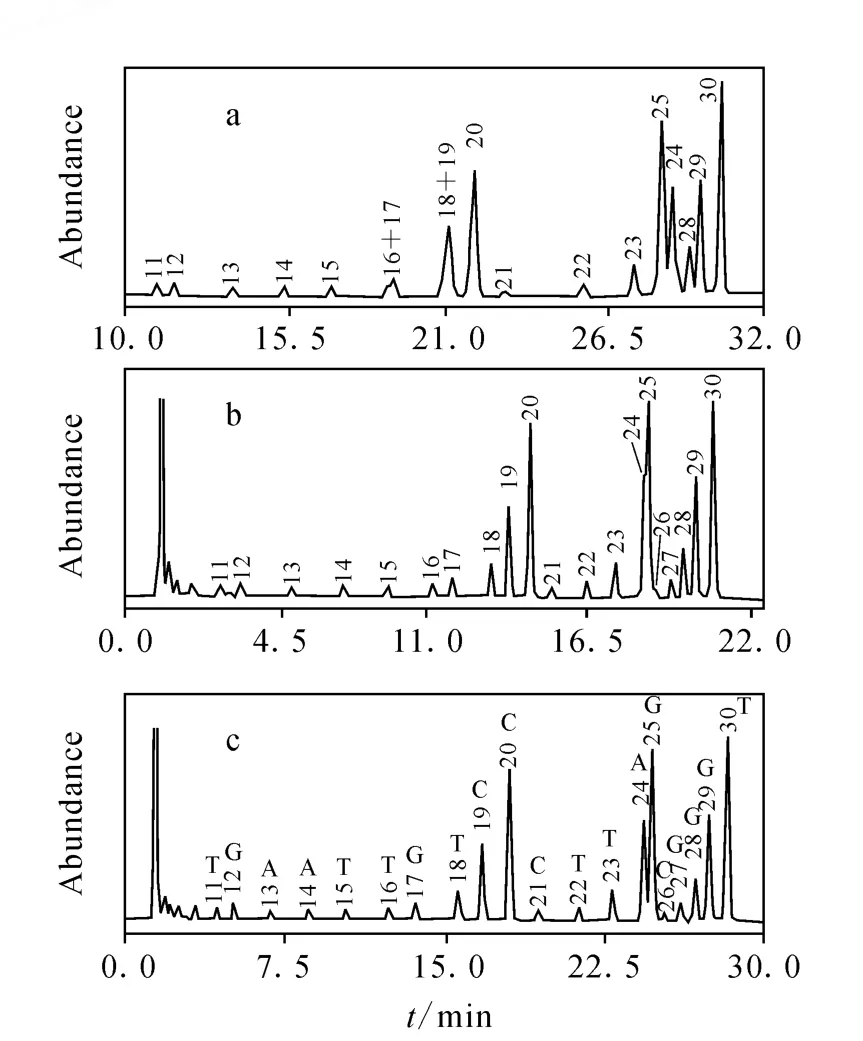
图5 使用不同离子对试剂缓冲液的核苷酸数10~30的寡核苷酸混合物分离效果对比[32]Fig.5 Separation of 10-30 mer oligonucleotides using different ion-pairing buffers[32]
在核酸水解酶的作用下,寡核苷酸能够在体内生成核苷酸片段断裂的代谢产物[38]。虽然质谱的选择反应监测(SRM)模式能够选择性监测原形和代谢产物,但由于同类寡核苷酸的碎片离子极为相似,在 SRM分析时可能发生交叉干扰[18-19],因此有必要提高色谱分离能力,避免原形和代谢物之间的干扰。目前,测定生物样品中寡核苷酸的液相色谱方法均采用梯度洗脱模式,与等度洗脱相比,这种色谱条件对寡核苷酸的分离度高,并且有助于改善色谱峰形、减少拖尾。但寡核苷酸对流动相中初始有机溶剂/水相比例很敏感,有机相/水相比例过小或过大会造成待测寡核苷酸不出峰或死时间出峰的极端结果。因此,在优化流动相梯度条件时需要谨慎选择初始有机溶剂/水相比例[32]。此外,使用缓慢的梯度变化条件也有助于原形和各种核苷酸片段水解的代谢产物实现基线分离[36]。
1.3 生物样品预处理
在复杂的生物基质中,存在大量盐类、磷脂、蛋白质和其他有机或者无机成分,对于小分子化合物而言,这些成分会对LC/MS分析产生影响并导致基质效应[39],但对于寡核苷酸这类生物大分子来说,基质中盐类的存在将导致大量金属加合离子的生成,而蛋白质则是影响其色谱行为并产生基质效应的主要因素。由于寡核苷酸经过结构改造后,蛋白结合率显著提高[40],因此在样品预处理阶段,去除生物样品中的盐和蛋白质对于LC/MS分析的顺利进行是至关重要的。沉淀蛋白法(PPT),固相萃取(SPE)和液液提取(LL E)均被应用于寡核苷酸的生物样品预处理。PPT法样品提取回收率低,并且由于难以充分去除蛋白质,在样品分析时会产生显著的基质效应[10];SPE法中应用比较广泛的是Oasis HLB柱(美国 Waters公司产品)[18-20,24],与色谱分离原理相同,在SPE小柱活化和淋洗的过程中均需要使用 TEA缓冲液才能达到保留寡核苷酸和洗脱干扰基质的目的,此外还有阴离子交换SPE柱 Poros HQ/M(美国 Perseptive Biosystems公司产品)[15,41]。但是,SPE法处理后的样品在LC/MS分析时也同样存在基质效应的影响[20,24]。苯酚-二氯甲烷/氯仿-异丙醇的 LL E法能够有效去除基质中的蛋白[25],SPE法能够富集待测物并进一步去除基质,这两种方法的结合使用不仅提取回收率较高,而且获得的样品在分析时还能充分避免基质的影响[18-19,42]。
寡核苷酸在聚丙烯管和玻璃容器中均存在非特异性吸附,对容器进行烷基化处理并采用含有 TEA的溶剂溶解样品,能够避免非特异性吸附[18]。在生物定量样品分析中,解决非特异性吸附的问题对保证结果的准确性和重现性也至关重要。
2 LC/MS方法应用于寡核苷酸药物的定量及定性研究
LC/MS法应用于寡核苷酸的生物样品定量分析列于表1。定量分析方法采用LLE、SPE或二者的组合进行样品预处理,液相色谱分离时多使用含有 TEA/HFIP的离子对色谱进行梯度洗脱,质谱检测均使用ESI源负离子的MRM扫描模式,并且将多组MRM信号加合,有助于提高质谱定量分析的灵敏度[19,23]。PF-ODN是辉瑞公司开发的用于治疗肺癌的含有24个核苷酸的PS-OGN类药物。Zhang等[18]建立了LCMS/MS法用于定量分析大鼠血浆中的原形化合物 PF-ODN,使用 Hypersil GOLD C18色谱柱,1.7 mmol/L TEA/100 mmol/L HFIP(p H 7.5)的流动相缓冲液体系梯度洗脱,定量下限为5.0μg/L。根据实际经验,Zhang等[18]提出了建立寡核苷酸类药物的LC/MS定量方法的一般流程,示于图6。本文作者[19]在此基础上进一步优化色谱和质谱条件,使用 Phenomenex GeminiC18色谱柱,流动相中加入2.85 mmol/L TEA/100 mmol/L HFIP(p H 7.8),在梯度洗脱条件下,使原形和4种核苷酸片段水解的代谢产物 5’N-1/3’N-1、5’N-2 和5’N-3基本实现色谱分离,避免了多组分在质谱检测中的相互干扰,实现了大鼠血浆中PF-ODN和4种代谢物的同时定量,定量下限为4.0 μg/L,示于图7。该方法被应用于大鼠静脉给予1.0 mg/kg PF-ODN后,血浆中原形及代谢物的定量,初步描述了 PF-ODN在大鼠体内的药动学过程。

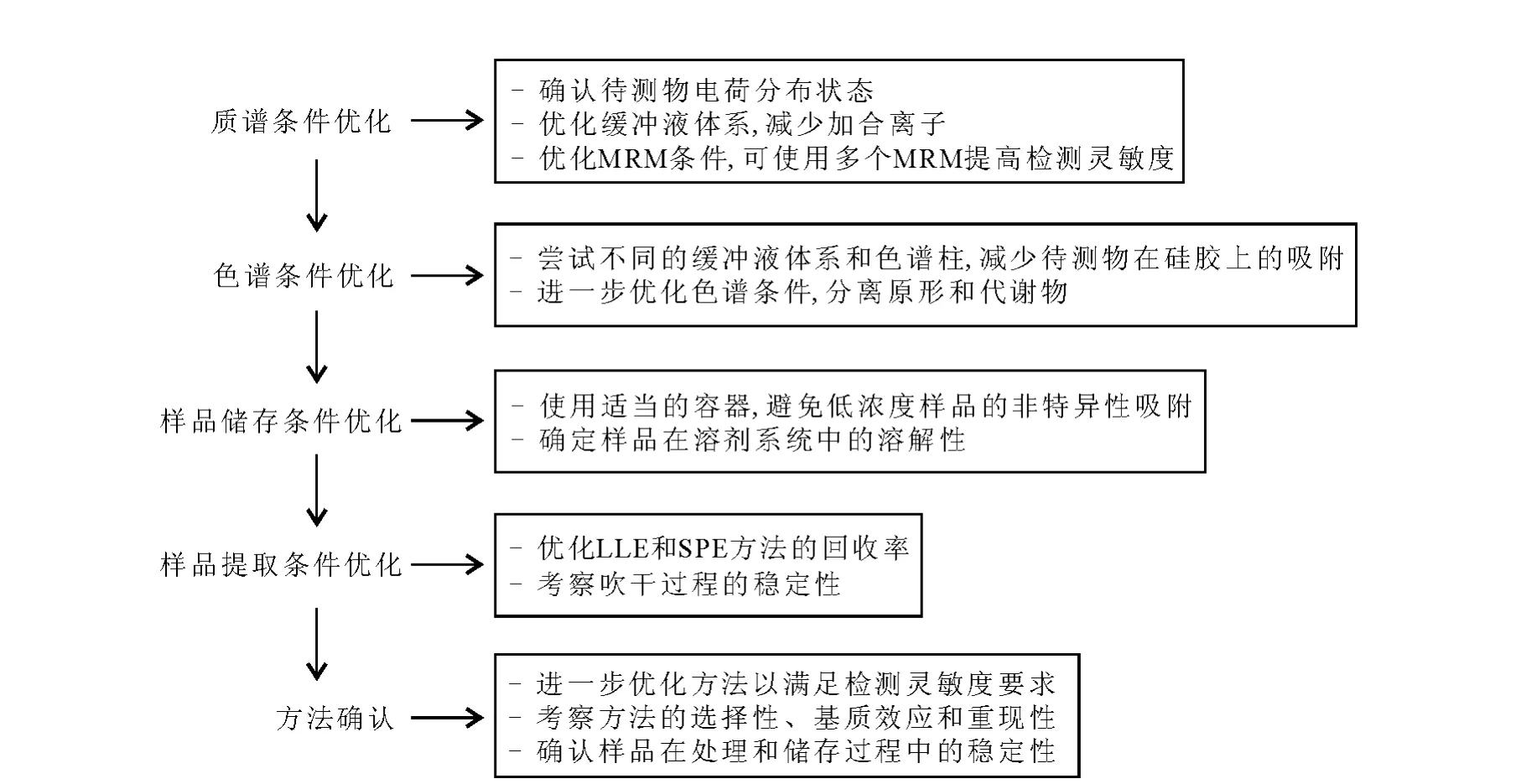
图6 寡核苷酸类药物的LC/MS生物分析方法建立流程Fig.6 Flowchart describes the recommended strategy for developing bioanalytical method of therapeutic oligonucleotides using LC/MS
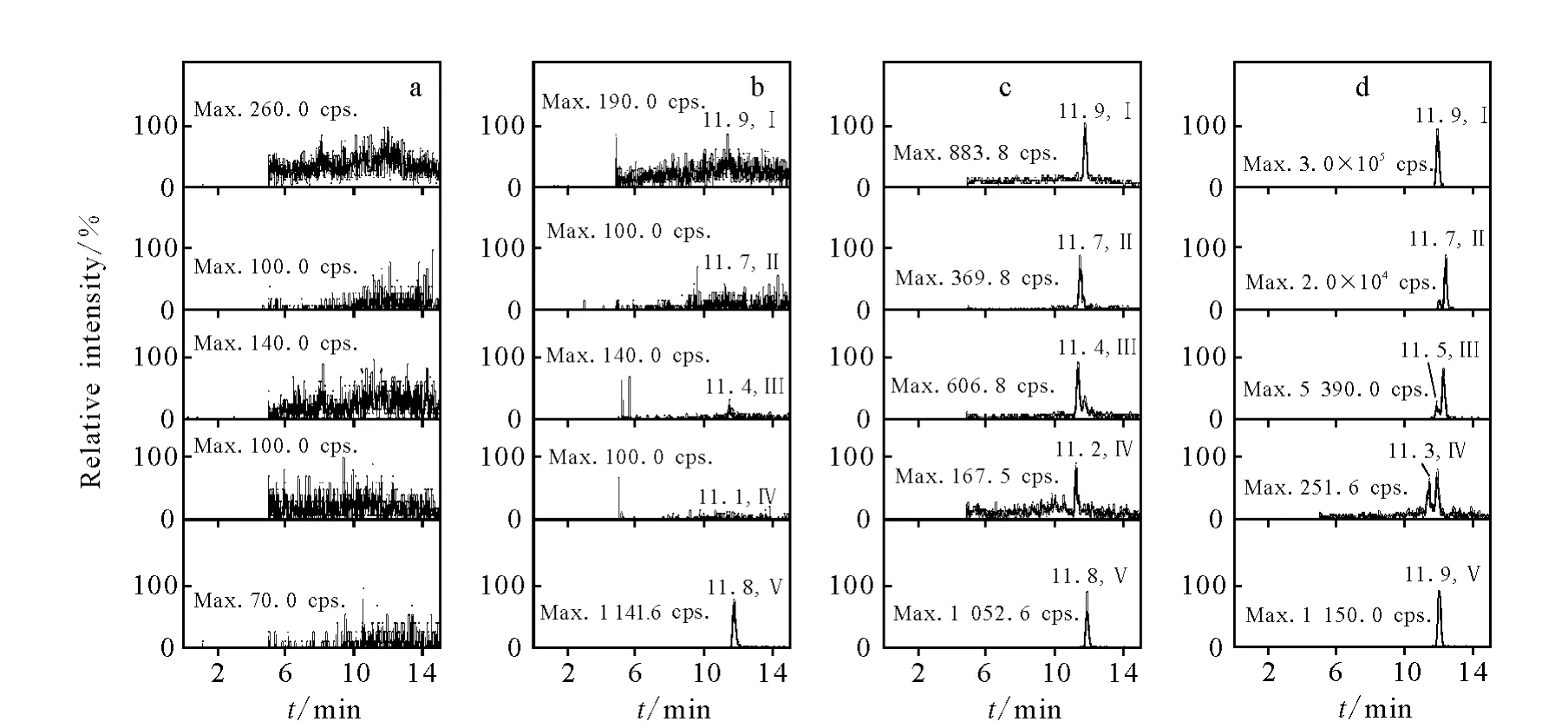
图7 大鼠血浆中PF-ODN及其代谢物的典型MRM色谱图Fig.7 Representative MRM chromatograms of PF-ODNand its metabolites in rat plasma
与小分子药物相同,成功的寡核苷酸药物需要有良好的体内代谢稳定性。确定寡核苷酸药物的代谢位点能够指导结构改造,了解代谢途径,有助于预测其毒性及药效。在体内,寡核苷酸可在3’-核酸外切酶和5’-核酸外切酶的作用下发生磷酸二酯键水解,分别从核酸链的3’和5’端丢失核苷酸片段。通常按照丢失的核苷酸数目将这些代谢产物标记为 3’N-1,5’N-1,3’N-2,5’N-2……等[19,27-28,43-44]。寡核苷酸代谢产物鉴定中常用的质谱仪是离子阱质谱,Wei等[27]使用离子阱质谱仪鉴定了 GTI-2040(5’-GGCTAAATCGCTCCACCAAG-3’) 在 不同生物样品中的代谢产物。在受试者血浆中共检测到5种 GTI-2040的3’端核苷酸依次水解的代谢产物,在大鼠肝肾匀浆液和人肝微粒体中也检测到3’端核苷酸水解的代谢产物。由于GTI-2040的3’和5’端均为鸟嘌呤核苷酸,根据准分子离子信息无法判断N-1(M1)这种代谢产物究竟是3’N-1还是 5’N-1,然而 ,离子阱质谱的优势在于能够获得w和a-B系列碎片离子,结合SOS软件可以确定相应的核苷酸序列。对比3’N-1和5’N-1对照品的二级质谱碎片离子信息发现,3’N-1和5’N-1对照品的w和a-B系列碎片离子存在明显区别,示于图8。图8a中,m/z674.9的w2-1离子和m/z870.4的a5-B5-2离子分别对应化合物 3’N-1的 3’AA和5’GGCTA核苷酸序列;而图 8b中,m/z691.1的w2-1离子和m/z862.6的a5-B5-2离子分别对应化合物5’N-1的 3’GA 和 5’GCTAA 核苷酸序列。根据这一特点,结合M1的二级质谱信息最终确认其为3’N-1核苷酸水解代谢产物。Zou等[30]使用L TQ Orbitrap高分辨质谱鉴定了 HBV263(siRNA)的体外代谢物,采集的LC/MS数据通过ProMass去卷积软件处理后,能够自动计算出代谢物的相对分子质量,并推测其可能的核苷酸序列,此外,高分辨的质谱数据能够准确区分质量数相差0.982 7 u的两个代谢物。
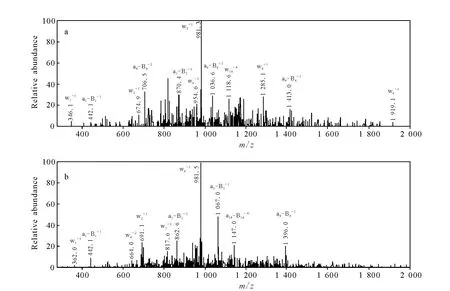
图8 m/z1 004[M-6H]6-的前体离子通过碰撞诱导解离产生的碎片离子及其归属[27]Fig.8 Fragment ions assignments following collision-induced dissociation of the precursor ions atm/z1 004[M-6H]6-[27]
3 总结和展望
随着LC/MS技术的发展,无论是在寡核苷酸类药物的生物样品定量分析方面,还是在代谢产物定性方面,LC/MS都具有显著的优势和鲜明的特点。因此,尽管这项分析技术目前还存在诸如进样器残留[19]、灵敏度不及 ELISA方法、样品预处理步骤繁琐等缺点,但它依然备受关注。LC/MS领域不断有新技术、新仪器被开发和使用,如专门针对消除残留设计的Nanospace HTS自动进样器(日本岛津公司产品);整合了新技术并使检测灵敏度显著提高的新型质谱仪,如Q-Trap 5500四极-线性离子阱杂交质谱仪(美国应用生物系统公司产品)、Xevo TQ三重四极杆串联质谱仪(美国沃特世公司产品)、TSQ Vantage三重四极杆串联质谱仪(美国赛默飞世尔公司产品)以及Agilent 6490三重四极杆串联质谱仪(美国安捷伦公司产品);适用于酸性强极性化合物的多种复合型SPE填料,以及用于寡核苷酸SPE快速提取的Clarity OTX试剂(美国菲罗门公司产品);根据亲水性相互作用开发的,适用于强极性化合物的多种 HILIC色谱柱[45]和专门针对寡核苷酸色谱分析开发的玻璃包覆棒状色谱柱(日本资生堂公司产品)的使用,可以提高LC/MS分析时流动相中有机相/水相的比例,有助于提高待测物离子化效率。这些技术在寡核苷酸类药物分析中的应用有待实践检验,相信LC/MS法在寡核苷酸生物样品分析领域的应用会不断深入。
[1]TREPANIER J B,TANNER J E,ALFIERI C.Oligonucleotide-based therapeutic options against hepatitis C virus infection[J].AntivirTher,2006,11(3):273-287.
[2]CROOKE S T,BENNETT C F.Progress in antisense oligonucleotide therapeutics[J].Annu Rev Pharmacol Toxicol,1996,36:107-129.
[3]WACHECK V.Oligonucleotide therapeutics-an emerging novel class of compounds[J].Wien Med Wochenschr,2006,156(17/18):481-487.
[4]SON TH EIM ER E J.Assembly and function of RNA silencing complexes[J].Nat Rev Mol Cell Biol,2005,6(2):127-138.
[5]LIN ZJ,LI W,DAI G.Application of LC-MS for quantitative analysis and metabolite identification of therapeutic oligonucleotides[J].J Pharm Biomed Anal,2007,44(2):330-341.
[6]AGRAWAL S.Importance of nucleotide sequence and chemical modifications of antisense oligonucleotides[J].Biochim Biophys Acta,1999,1 489(1):53-68.
[7]L EVIN A A.A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides[J].Biochim Biophys Acta,1999,1 489(1):69-84.
[8]MCKAY R A,MIRAGLIA L J,CUMMINS L L,et al.Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression[J].J Biol Chem,1999,274(3):1 715-1 722.
[9]KARKARE S,BHATNAGAR D.Promising nucleic acid analogs and mimics:characteristic features and applications of PNA,LNA,and morpholino[J].Appl Microbiol Biotechnol,2006,71(5):575-586.
[10]YU R Z,GEARY R S,L EVIN A A.Application of novel quantitative bioanalytical methods for pharmacokinetic and pharmacokinetic/pharmacodynamic assessments of antisense oligonucleotides[J].Curr Opin Drug Discov Devel,2004,7(2):195-203.
[11]DEV ERRE J R,BOU TET V,BOQU ET D,et al.A competitive enzyme hybridization assay for plasma determination of phosphodiester and phosphorothioate antisense oligonucleotides[J].Nucleic Acids Res,1997,25(18):3 584-3 589.
[12]YU R Z,BAKER B,CHAPPELL A,et al.Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma[J].Anal Biochem,2002,304(1):19-25.
[13]杨何义,蔡 耘,钱小红.生物质谱在核糖核酸领域的应用[J].质谱学报,2004,25(1):52-60.
[14]HUBER C G,OBERACHER H.Analysis of nucleic acids by on-line liquid chromatography-mass spectrometry[J].Mass Spectrom Rev,2001,20(5):310-343.
[15]GAUS H J,OWENS S R,WINNIMAN M,et al.On-line HPLC electrospray mass spectrometry of phosphorothioate oligonucleotide metabolites[J].Anal Chem,1997,69(3):313-319.
[16]APFFEL A,CHAKEL J A,FISCHER S.Analysis of oligonucleotides by HPLC-electrospray ionizaiton mass spectrometry[J].Anal Chem,1997,69(7):1 320-1 325.
[17]MURPH Y A T,BROWN-AU GSBURGER P,YU R Z,et al.Development of an ion-pair reverse-phase liquid chromatographic/tandem mass spectrometry method for the determination of an 18-mer phosphorothioate oligonucleotide in mouse liver tissue[J].Eur J Mass Spectrom(Chichester,Eng),2005,11(2):209-215.
[18]ZHANG G,LIN J,SRINIVASAN K,et al.Strategies for bioanalysis of an oligonucleotide class macromolecule from rat plasma using liquid chromatography-tandem mass spectrometry[J].Anal Chem,2007,79(9):3 416-3 424.
[19]DENG P,CHEN X,ZHANG G,et al.Bioanalysis of an oligonucleotide and its metabolites by liquid chromatography-tandem mass spectrometry[J].J Pharm Biomed Anal,2010,52(4):571-579.
[20]DAI G,WEI X,LIU Z,et al.Characterization and quantification of Bcl-2 antisense G3139 and metabolites in plasma and urine by ion-pair reversed phaseHPLC coupled with electrospray ion-trap mass spectrometry[J].J Chromatogr B,2005,825(2):201-213.
[21]CRAIN P F,MCCLOSKEYJ A.Applications of mass spectrometry to the characterization of oligonucleotides and nucleic acids[J].Curr Opin Biotechnol,1998,9(1):25-34.
[22]DEGUCHI K,ISHIKAWA M,YOKOKURA T,et al.Enhanced mass detection of oligonucleotides using reverse-phase high-performance liquid chromatography/electrospray ionization ion-trap mass spectrometry[J].Rapid Commun Mass Spectrom,2002,16(22):2 133-2 141.
[23]XU K,WILLIAMS W A,WONG S,et al.Quantitation of antisense oligonucleotides in mouse plasma by LC-MS/MS[C]//Proceedings of the 51st ASMS Conference on Mass Spectrometry and Allied Topics,Montreal,Canada,2003.
[24]JOHNSON J L,GUO W,ZANGJ,et al.Quantification of raf antisense oligonucleotide(rafAON)in biological matrices by LC-MS/MS to support pharmacokinetics of a liposome-entrapped rafAON formulation[J].Biomed Chromatogr,2005,19(4):272-278.
[25]HE L,CHEN Y S,LAM Z,et al.Antisense oligonucleotides(ASO)metabolism study by LC/MS/MS in support of ASO drug discovery and development[C]// Proceedings ofthe52nd ASMS Conference on Mass Spectrometry and Allied Topics,Nashvile,TN,2004.
[26]MCLUCKEY S A,GARYJ V B,GLISH G L.Tandem mass spectrometry of small,multiply charged oligonucleotides[J].JAm Soc Mass Spectrom,1992,3:60-70.
[27]WEI X,DAI G,L IU Z,et al.Metabolism of GTI-2040,a phosphorothioate oligonucleotide antisense,using ion-pair reversed phase high performance liquid chromatography(HPLC)coupled with electrospray ion-trap mass spectrometry[J].AAPS J,2006,8(4):E743-E755.
[28]BEVERLY M,HARTSOUGH K,MACHEMER L,et al.Liquid chromatography electrospray ionization mass spectrometry analysis of the ocular metabolites from a short interfering RNA duplex[J].J Chromatogr B,2006,835(1/2):62-70.
[29]ROZENSKIJ,MCCLOSKEYJ A.SOS:A simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry[J].J Am Soc Mass Spectrom,2002,13(3):200-203.
[30]ZOU Y,TILL ER P,CHEN I W,et al.Metabolite identification of small interfering RNA duplex by high-resolution accurate mass spectrometry[J].Rapid Commun Mass Spectrom,2008,22(12):1 871-1 881.
[31]GILAR M.Analysis and purification of synthetic oligonucleotides by reversed-phase high-performance liquid chromatography with photodiode array and mass spectrometry detection[J].Anal Biochem,2001,298(2):196-206.
[32]GILAR M,FOUN TAIN K J,BUDMAN Y,et al.Ion-pair reversed-phase high-performance liquid chromatography analysis of oligonucleotides:retention prediction[J].J Chromatogr A,2002,958(1/2):167-182.
[33]BOTHNER B,CHATMAN K,SARKISIAN M,et al.Liquid chromatography mass spectrometry of antisense oligonucleotides[J].Bioorg Med Chem,1995,5(23):2 863-2 868.
[34]GILAR M,BOUVIER E S P.Purification of crude DNA oligonucleotides by solid-phase extraction and reversed-phase high-performance liquid chromatography[J].J Chromatogr A,2000,890(1):167-177.
[35]NORDHOFF E,KIRPEKAR F,ROEPSTORFF P.Mass spectrometry of nucleic acids[J].Mass Spectrom Rev,1996,15:67-138.
[36]GILAR M,NEUE U D.Peak capacity in gradient reversed-phase liquid chromatography of biopolymers.Theoretical and practical implications for the separation of oligonucleotides[J].J Chromatogr A,2007,1 169(1/2):139-150.
[37]FOUNTAIN KJ,GILAR M,GEBLER J C.A-nalysis of native and chemically modified oligonucleotides by tandem ion-pair reversed-phase highperformance liquid chromatography/electrospray ionization mass spectrometry[J].Rapid Commun Mass Spectrom,2003,17(7):646-653.
[38]AGRAWAL S,ZHANG R.Pharmacokinetics of oligonucleotides[J].Ciba Found Symp,1997,209:60-75.
[39]GOSETTI F,MAZZUCCO E,ZAMPIERI D,et al.Signal suppression/enhancement in highperformance liquid chromatography tandem mass spectrometry[J].J Chromatogr A,2010,1 217(25):3 929-3 937.
[40]DEAN N M,BUTL ER M,MONIA B P,et al.Antisense drug technology:Principles,strategies,and applications[M].New York:Marcel Dekker,2008:187.
[41]YU R Z,GEARY R S,MONTEITH D K,et al.Tissue disposition of2’-O-(2-methoxy)ethyl modified antisense oligonucleotides in monkeys[J].J Pharm Sci,2004,93(1):48-59.
[42]GEARY R S,YU R Z,WATANABE T,et al.Pharmacokinetics of a tumor necrosis factor-alpha phosphorothioate 2’-O-(2-methoxyethyl)modified antisense oligonucleotide:comparison across species[J].Drug Metab Dispos,2003,31(11):1 419-1 428.
[43]GRIFFEY R H,GREIG M J,GAUS H J,et al.Characterization of oligonucleotide metabolism in vivo via liquid chromatography/electrospray tandem mass spectrometry with a quadrupole ion trap mass spectrometer[J].J Mass Spectrom,1997,32(3):305-313.
[44]BEVERL Y M,HARTSOU GH K,MACHEMER L.Liquid chromatography/electrospray mass spectrometric analysis of metabolites from an inhibitory RNA duplex[J].Rapid Commun Mass Spectrom,2005,19(12):1 675-1 682.
[45]DENG P,CHEN X Y,ZHONGD F.Quantification of polar drugs in human plasma with LC-MS/MS[J].Bioanalysis,2009,1(1):187-203.
Quantitative Analysis and Metabolite Identification of Therapeutic Oligonucleotides in Biological Samples Using LC/MS
DENG Pan,ZHONG Da-fang,CHEN Xiao-yan
(S hanghai Institute of Materia Medica,Chinese Academy of Sciences,Shanghai201203,China)
Therapeutic oligonucleotides have become as a new drug class.They are synthetic DNA or RNA that typically between 15 and 50 nucleotide units long.In the pharmacokinetic studies of therapeutic oligonucleotides,the classical method was enzyme-linked immunosorbent assay(ELISA).However,the development of ELISA method was time-consuming and expensive,and has become the bottleneck in the new drug research and development.The technical issues associated with liquid chromatography-mass spectrometry(LC/MS)method in the bioanalysis of oligonucleotides included:limited ionization efficiency,severe matrix effect,incompatibility of chromatographic condition and MS detection,and low sample preparation recovery.However,the development of an LC/MS method was fast,and this method was selective.This review summarized the development and applications of LC/MS method for the quantitation of therapeutic oligonucleotides and characterization of their metabolites in biological samples described in the literatures over the past 15 years.
therapeutic oligonucleotides;liquid chromatography-mass spectrometry(LC/MS);biological samples
陈笑艳(1971~),女,黑龙江人,研究员,从事药物代谢与药代动力学研究。E-mail:xychen@mail.shcnc.ac.cn
O 657.63
A
1004-2997(2011)01-0013-11
2010-12-04;
2011-01-14
邓 泮(1984~),女,陕西人,博士研究生,从事药物代谢与药代动力学研究。E-mail:panda4177@126.com
猜你喜欢
化工设计通讯(2022年6期)2023-01-02
现代临床医学(2022年4期)2022-09-29
世界科学技术-中医药现代化(2022年3期)2022-08-22
北京生物医学工程(2022年4期)2022-08-18
肝博士(2022年3期)2022-06-30
中国医院用药评价与分析(2020年3期)2020-05-29
农药科学与管理(2019年5期)2019-08-13
中国饲料(2019年19期)2019-03-25
生物工程学报(2018年5期)2018-06-11
