
水溶性五环三萜酸衍生物的制备及研究进展
2011-02-02 06:16尚娟陈莉
药学进展 2011年5期
尚娟,陈莉
(中国药科大学天然药物化学教研室,江苏南京 210009)
水溶性五环三萜酸衍生物的制备及研究进展
尚娟,陈莉*
(中国药科大学天然药物化学教研室,江苏南京 210009)
综述几种常见水溶性五环三萜酸衍生物的研究进展。五环三萜酸类化合物多具有广泛的生物活性,但因水溶性差、生物利用度低使其临床应用受到限制。以五环三萜酸为先导化合物,经结构修饰合成水溶性衍生物,并从中寻找出有生物活性和临床应用价值的化合物是当前天然药物化学研究的热点之一。
五环三萜酸;水溶性衍生物;生物活性
五环三萜酸类化合物广泛存在于自然界多种植物中。常见的五环三萜酸有齐墩果烷型三萜酸,如齐墩果酸(oleanolic acid,OA,1)、甘草次酸(glycyrrhetinic acid,GA,2)和山楂酸(masilinic acid,MA,3)等,乌苏烷型三萜酸乌苏酸,又名熊果酸(ursolinicacid,UA,4)及羽扇豆烷型三萜酸如白桦酸(betulinic acid,BA,5)等。
上述三萜酸均具有多种生物活性,如OA除具有显著的保肝作用外,还有抗氧化、抗肿瘤活性及降糖作用[1-4];GA具有保肝、降血脂、降血糖、肾上腺皮质激素样作用[5-9];MA除有抗肿瘤等活性外,还可显著对抗肾上腺素和葡萄糖引起的血糖升高[10-11];UA具有抗肿瘤、抗病毒和抗菌活性以及保肝、调节中枢神经系统等作用[12-14];BA则具有抗肿瘤、抗HIV、抗炎、抗菌、提高机体免疫力等生物活性,其中以抗肿瘤和抗HIV活性最为突出[15-16]。此外,三萜酸类化合物使用安全且价廉易得,因此,该类化合物受到了越来越广泛的关注。目前,已有数个三萜酸用于临床或进入临床试验,如OA作为治疗急性黄疸性肝炎和慢性肝炎的非处方药已在临床应用多年;BA已进入Ⅱ期临床用于黑色素瘤的治疗;GA作为肾上腺皮质激素类药物,临床上可代替去氧皮质酮用于慢性肾上腺皮质功能减退症及眼睛等部位的多种炎症。然而,五环三萜酸类化合物因具有刚性骨架和强疏水性结构,导致水溶性差,从而影响了其在胃肠道中的溶出和吸收,生物利用度低,严重制约了其临床应用。为此,药学工作者采用各种化学方法,如通过成盐、以共价键键合亲水性基团和形成水溶性复合物等来改善五环三萜酸的水溶性。本文对近年来该领域的研究进展作一综述。

1 成盐
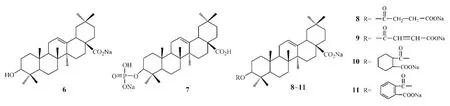
成盐可显著改善五环三萜酸的水溶性。例如,赵争胜等将OA制成钠盐(6),后者在水中的溶解度为OA的27倍(中成药,1991年);徐晓娣等[17]将该OA钠盐与水飞蓟素制成复方软胶囊给大鼠灌胃使用,结果发现,其相对生物利用度达194.66%。
研究表明:将药物与磷脂反应形成的药物磷脂衍生物制成钠盐可使药物的溶解度等理化性质发生显著变化[18],从而促进药物吸收。例如,刘明生等将OA与三氯氧磷作用生成其磷酸酯,并制成单钠盐(7),结果显示,该钠盐的抗肿瘤活性为OA的 5倍(中国药物化学杂志,1993年)。
蒋朝晖等[19]将OA的C3位羟基与二元酸形成单酯,并制成二钠盐,得到4个衍生物(8~11)。结果,在25℃下,化合物8~11在水中的溶解度依次为11.2、18.5、10.4和33.3 g·L-1;而相同条件下,OA的溶解度仅为4.61 mg·L-1。初步动物体内实验结果表明:与OA相比,化合物8~11对实验性肝损伤的保护作用均有不同程度的提高,其中化合物11的刺激性和毒性较小,且稳定性较好,其对大、小鼠实验性肝损伤(D-Galn、CCl4、CdCl2致肝损伤)的保护作用显著强于OA,量效关系呈正相关,且无OA在大剂量时会加重动物肝脏损伤的副作用。
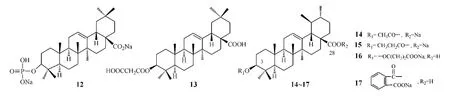
至今未见有OA在人体内的生物利用度的具体数据报道,原因可能为其生物利用度低,有关药动学研究也较少。王文宇等[20]采用HPLC法测定了大鼠血浆中OA水溶性衍生物齐墩果酸磷酸酯二盐(12)的浓度,并对其在大鼠体内的药动学和生物利用度进行了研究。结果显示,将化合物12经灌胃及肝门静脉给予大鼠后,其药时过程符合单室开放一级吸收模型特征;经灌胃、肝门静脉、颈静脉交叉给药后,灌胃的绝对生物利用度为22.0%,肝门静脉注射的绝对生物利用度则达88.89%。
有研究称,将OA进行磷酸酯化后制成齐墩果酸磷酸酯单钠盐,其抗肿瘤活性为OA的5倍(刘明生等,中国药物化学杂志,1993年),Ma等[21]也发现,在OA的C3位羟基引入二羧酸形成的二羧酸半酯能使OA的抗肿瘤活性大大提高,其中衍生物13对HCV蛋白酶的IC50为9 mg·L-1,而OA的IC50为26 mg·L-1。据此,艾宏儒等[22]对UA的C3位进行了结构修饰,合成了一系列二羧酸半酯衍生物并将其制成钠盐(14~17)。结果表明:化合物14~17的水溶性强于UA,且在水中和酸性条件下均很稳定,因此可研究将其制成注射液或肠溶口服制剂。但该文未报道上述衍生物的抗肿瘤活性数据。
含氮化合物广泛存在于自然界,许多有机含氮化合物具有显著的生物活性,如生物碱。在三萜酸类化合物中引入氮原子常能增加化合物的抗肿瘤和抗病毒活性,且其中某些含氮化合物与酸成盐后溶解性明显增加。例如,申利红等[23]合成了一系列一氧化氮供体型硝酸酯类GA衍生物,为改善衍生物的溶解性,他们先用氯乙酰氯在C3位羟基上酯化,然后再与仲胺反应,最后与盐酸成盐,得到12个目标化合物,抗肿瘤活性初筛结果显示,硝酸酯类GA衍生物的C3位引入碱性基团并制成相应的盐后,抗肿瘤活性均明显提高,其中化合物18对MCF-7、人早幼粒细胞白血病细胞HL-60、人肝癌细胞HepG2和BEL-7402的IC50均低于GA(7.56vs9.01,7.60vs47.79,4.56vs4.61,5.28vs5.75μmol·L-1),故可作为较有潜力的抗肿瘤先导化合物。

2 偶联亲水性分子
2.1 偶联氨基酸
氨基酸类化合物在体内有特定的转运系统,且其本身水溶性好,在药物分子结构中引入氨基酸基团,可增加药物的水溶性,从而促进吸收,提高生物利用度。如Yoshimi等发现,在难溶性药物分子中引入氨基酸分子制备成水溶性前药,可使药物的吸收显著增加(JPharmacobiod,1992年)。
Li等[24]通过先将OA的A环氧化或偶联含氮杂环(吲哚、吡嗪和喹啉),再与氨基酸偶联的方法得到一系列衍生物,并测定了这些化合物对1α,25-二羟维生素D3诱导的酒石酸酸性磷酸酶(TRAP)阳性破骨细胞样多核细胞(OCL)形成的抑制活性:将OCL分别与0.01μmol·L-1的1α,25-二羟维生素D3及浓度为0.2μmol·L-1的化合物19和20共培养,结果两组的细胞形成率分别为31.7%和34.1%,而与浓度为2μmol·L-1的OA和0.01μmol·L-1的1α,25-二羟维生素D3共培养组的细胞形成率为89.5%(以仅用0.01μmol·L-1的1α,25-二羟维生素D3时的细胞形成率为100%计,能使细胞形成率低于75%的化合物即被认为具有潜在活性)。
构效关系研究表明:偶联物中氨基酸链越短则活性越强,如与OA衍生物和甘氨酸的偶联物共培养组OCL的形成率低于OA衍生物和丙氨酸的偶联物组(34.1%vs77.0%)。
有文献报道,OA经氧化得到的3-氧代OA的抗肿瘤活性明显提高,但水溶性差(最大溶解量低于1 mg·L-1)[25],而当BA与一系列氨基酸连接后可提高水溶性,并保持细胞毒性[26]。据此,孙华等[25]设计合成了9个3-氧代OA氨基酸偶联物,并用HPLC法和MTT法分别测定了这些偶联物的水溶性及对人口腔鳞癌KB细胞和人口腔鳞癌长春新碱耐药株KB/V的体外抑制活性,结果发现,与3-氧代OA相比,有6个偶联物的水溶性有不同程度的改善,其中化合物21的水溶性最好,其在水中的最大溶解量为9.067 mg·L-1;水溶性较高的偶联物21~23在浓度为10μmol·L-1时,对KB细胞的抑制率分别为10.17%、10.47%和3.65%,对KB/V细胞的抑制率分别为12.15%、2.43%和-2.48%[肿瘤细胞生长抑制率=(1-实验组OD)/对照组OD×100%,OD为酶标仪上于570 nm波长处测得的光密度值]。而文献[27]报道3-氧代OA对KB和KB/V细胞的IC50分别为14.3和25.5μmol·L-1。

Meng等[28]合成了C28位偶联丝氨酸、甘氨酸和苯丙氨酸等的UA衍生物,以增加其水溶性,在测定它们对肿瘤的抑制活性时发现,这些衍生物对HeLa细胞的抑制活性均强于UA,其中化合物24对Hela细胞、人卵巢癌细胞SKOV3和人胃癌细胞BGC-823的IC50分别为2.62、2.24和8.30μmol·L-1,而UA对这3种细胞的IC50均大于10μmol·L-1。
BA的C3位羟基、C28位羧基和C19位烯丙基均可作为修饰位点,Evers等根据生物电子等排原理,在BA的C19位用硫醚基团取代烯丙基得到了一系列具有抗HIV活性的BA衍生物(JMedChem,1996年)。而Qian等[29]亦设计合成了17个C28,30-双取代的BA衍生物和7个C3,28-双取代的BA衍生物,对这些衍生物的抗HIV-1增殖活性及水溶性进行的测定表明:化合物25对HIV-1Ⅲ感染者的白血病细胞MT-2的EC50为0.09μmol·L-1,与HIV侵入抑制剂IC9564(26)相当,但水溶性比后者好(因化合物26的C19位的烯丙基不能与水形成氢键,而化合物25的C30位连接了亲水性基团可与水形成氢键)。此外,他们还发现,环状仲胺形成的C28位酰胺键可明显增强衍生物在人肝微粒体中的代谢稳定性,其中的C3,28-双取代的BA衍生物(27,28)的EC50可达7和6 nmol·L-1,活性与已进入Ⅱ期临床研究的bevirimat(2,3',3'-二甲基琥珀酰白桦酸)(EC50为7 nmol·L-1)相当,可望开发成代谢稳定的新一代HIV抑制剂。


BA对多种癌细胞株均有细胞毒性,但由于其在水中几乎不溶而影响了吸收。为此,Drag-Zalesinska等[30]将BA与各种氨基酸反应生成一系列单取代或双取代的BA-氨基酸酯,这些衍生物的水溶性均大大提高,并可在进入体内后再水解成BA和氨基酸。其中水溶性最好的是化合物29,活性也最强,对人胰腺癌细胞株EPP85-181P、人胰腺癌柔红霉素耐药株EPP85-181RDB和人胃癌细胞株EPG85-257P的IC50分别为3.8、18和4.1μmol·L-1,而BA对这3种细胞的IC50均大于20μmol·L-1。
Jeong等[26]将BA的C28位羧基与一系列氨基酸偶联,并测定这些BA-氨基酸酯对人黑色素瘤MEL-2细胞和KB细胞株的活性。结果表明:与BA相比,BA-氨基酸酯不仅水溶性提高,对癌细胞的选择性也得到增强。其中化合物30的水溶性为BA的10倍,其对MEL-2细胞和KB细胞的ED50分别为1.5、4.6 mg·L-1,而BA对这两种细胞的ED50分别为4.2及20 mg·L-1以上。

Parra等[31]采用液相和固相合成法在MA的C28位羧基上偶联α-或ω-氨基酸,合成了一系列带单个氨基酸残基的络合物及二肽、三肽化合物,这些化合物的水溶性增加,并且对HIV有明显的抑制活性:在用荧光法测定与这些化合物共培养的MT-2细胞(用重组的能表达荧光素酶的HIV-1感染,病毒增殖越快则荧光越强)的荧光变化时,10μmol·L-1的化合物31和32组细胞的荧光强度分别降至空白组的32.5%和44.9%,而MA(10μmol·L-1)组则为56.3%;进一步实验发现,化合物31、32对感染HIV-1的MT-2细胞的细胞周期无影响,而MA则对之有影响;此外,化合物31、32和MA一样可诱导该MT-2细胞的凋亡,在浓度为10μmol·L-1时,各组细胞的凋亡率分别为3.5%、2.1%和13.5%;在浓度为25μmol·L-1时,凋亡率分别为3.2%、2.6%和51.8%。因此,与MA相比,化合物31、32的细胞毒性更低,有望开发成一类新型的HIV抑制剂。

2.2 制成糖苷
五环三萜酸与糖成三萜苷后,羟基数目增多,极性增大,水溶性大大改善。其实,引入糖基不仅可增加水溶性和细胞渗透性,某些特定的糖基的引入还可增加靶向性,如引入半乳糖可增加肝靶向性,此外引入的糖基可通过与细胞内/细胞间糖类-蛋白质的相互作用以增强对靶细胞的选择性。
臧静等[32]在OA的C3位羟基上进行糖苷化反应,合成了4种专一的立体选择性的OA皂苷,并初步探讨了糖链的结构对此类化合物抗肿瘤活性的影响。结果显示,连有半乳糖和阿拉伯糖的OA皂苷(化合物33、34)对P388小鼠白血病细胞株和A549人肺癌细胞株表现出较弱的抑制活性,其在浓度为0.01μmol·L-1时对P388细胞的抑制率分别为13.6%和4.6%;对A549细胞的抑制率分别为19.7%和19.4%,而其它化合物则无效,表明不同的糖基对抗肿瘤活性的影响不同。
Wang等[33]合成了14个N-乙酰基-β-D-葡萄糖胺的OA皂苷,并测试了这些化合物对3种细胞株的细胞毒性。结果显示,化合物OA-3-O-β-D-葡萄糖乙酰胺(35)在10μmol·L-1浓度下对HL-60、BGC-823和HeP-2细胞株的抑制率分别为82.62%、68.99%和23.34%,而相同浓度的OA对这3种细胞株的抑制率分别为13.18%、17.00%和26.99%。

OA-NO供体衍生物(36)在体内外对人肝癌细胞均有很强的细胞毒性(如对HepG2细胞的IC50为1.37μmol·L-1),但其水溶性很差(当pH为6.5~7.8时,其在水中的最大溶解量不超过1 mg·L-1),即使将其制成微乳和脂质体也未能增加水溶性[34]。Huang等[34]将糖分子引入化合物36,得到其一系列糖苷衍生物,其中包括化合物37。体外实验表明:衍生物37对人肝癌细胞HepG2、SMMC-7721和BEL-7402有细胞毒活性,IC50分别为2.13、1.18、2.96μmol·L-1,而化合物36的IC50分别为1.37、4.78、14.5μmol·L-1。用移植SMMC-7721的小鼠进行的体内实验表明:衍生物37(25 mg·kg-1, iv)可显著抑制癌细胞的生长,其活性强于5-FU (25 mg·kg-1,iv)(IC50,1.18vs43.5μmol·L-1),且给药21天后,衍生物37组小鼠的实体瘤质量明显低于空白对照组(0.31vs0.87 g,P<0.01)。提示化合物37可作为治疗肝癌的候选药物。而且,衍生物37的水溶性明显增加,在甲基乙二醇和5%聚乙二醇15羟基硬脂酸水溶液中的最大溶解量高于化合物36,分别为16.2vs2.23,0.826vs0.158 g·L-1。据此,Huang等以5%甲基乙二醇、5%聚乙二醇15羟基硬脂酸和5%无水乙醇的水溶液为溶剂,将衍生物37制成5 g·L-1的注射液用以评价其体内活性。

28 -O-β-D-BA葡萄糖醛酸苷(38)是BA在体内的主要代谢产物之一,水溶性优于BA,且其可在β-D-葡萄糖醛酸酶存在下水解出具有抗癌活性的BA,且β-D-葡萄糖醛酸酶在坏死的肿瘤组织内高度表达。Gauthier等[35]在相转移条件下,用2,3,4-三-O-乙酰基-1-溴-D-吡喃葡萄糖醛酸甲酯首次合成了化合物38,并测定了其在体外磷酸盐缓冲液(0.02mol·L-1,pH 7.2)中于37℃下放置1周的稳定性、溶血性和水溶性。结果显示,化合物38在上述条件下放置后无降解产物生成,对A549细胞、人直肠结肠腺癌细胞DLD-1和人胚胎皮肤细胞WS1的IC50均大于100μmol·L-1,而对绵羊红细胞的半数溶血剂量(HD50)大于100μmol·L-1。
研究发现,抑制糖原磷酸化酶(GP)的活性对控制血糖治疗糖尿病有重要作用,而葡萄糖的类似物可通过与GP的催化位点结合而发挥对GP的抑制活性。Cheng等[36]在OA的C3、C28位通过三唑基团或酯键与葡萄糖偶联,合成了一系列OA糖苷衍生物,其对兔肌肉GP的抑制活性测定结果显示,与OA相比,化合物39对GP的抑制活性更强(IC50,1.14vs14μmol·L-1)。构效关系研究表明:糖基与三萜之间的连接基团越短,对GP的抑制活性越强;连接基团的亲水性越强,对GP的抑制活性也越强;连接基团连接在OA的C28位时对活性的影响比连在C3位时大。

3 形成水溶性复合物
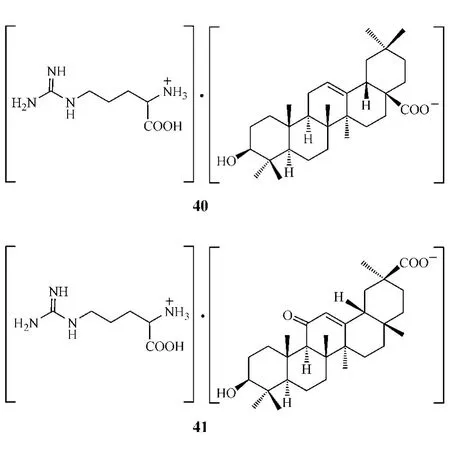
L-精氨酸为碱性氨基酸,可与三萜酸形成复合物。张丽娟等[37]分别采用超声法、搅拌法和回流法制备了L-精氨酸OA复合物(40);许卉等[38]则采用搅拌法制备了精氨酸GA复合物(41)。形成的复合物水溶性比OA和GA好,可制成适合临床使用的剂型,同时还可降低血管痉挛的发生,从而提高用药安全性。
OA具有强疏水性,影响了在胃肠道中的溶出和吸收;而磷脂具有乳化、分散、助渗、润湿等特性,并与细胞表面有较强的亲和性,此外,磷脂还可提高血液胆固醇水平,并对肝脏有一定的保护作用。因此,将OA与磷脂形成复合物,有望增强活性,提高稳定性,促进吸收,提高生物利用度,并可发挥二者在肝病治疗上的协同作用。马金玲[39]制备了OA与大豆磷脂及蛋黄磷脂等的OA磷脂复合物。溶出度实验显示,OA磷脂复合物的溶出度远大于OA (87.7%vs27.3%)。
4 结语
五环三萜酸是天然产物中广泛存在的一类化合物,其活性多样。近年来,关于五环三萜酸的结构修饰和改造的文献报道较多,其中也得到了一些活性较好的衍生物,但水溶性差仍是主要问题。因此,在五环三萜酸的结构修饰和改造中,关键是如何在保持或提高生物活性的前提下改善其水溶性。很多五环三萜酸中都有—OH和—COOH基团,在这两个位点上引入极性基团,可有效改善分子的水溶性,在本文中提到的3种方法中,偶联亲水性分子法由于连接上的基团与机体的生物相容性较好或本身就是天然产物而具有更好的研究前景。除这3种方法外,在—OH和—COOH上连接除氨基酸和糖基外的溶解性较好且可增强其活性的其它分子或基团也是值得研究的方向。笔者所在课题组以齐墩果酸、乌苏酸为先导化合物,已设计并合成了一系列水溶性呋咱类和硝酸酯类一氧化氮供体型齐墩果酸和乌苏酸衍生物,相关结构确证和活性筛选工作正在进行之中。
[1]Reisman S A,Aleksunes L M,Klaassen C D.Oleanolic acid activates NrF2 and protects from acetaminophen hepatotoxicity via NrF2-dependent and NrF2-independent processes[J].Biochem Pharmacol,2009,77(7): 1273-1282.
[2]Teodoro T,Zhang L,Alexander T,et al.Oleanolic acid enhances insulin secretion in pancreaticβ-cells[J].FEBS Lett,2008,582(9):1375-1380.
[3]Braga F,Ayres-Saraiva D,Gattass C R,et al.Oleanolic acid inhibits the activity of the multidrug resistance protein ABCC1(MRP1)butnot of the ABCB1(P-glycoprotein):possible use in cancer chemotherapy[J].Cancer Lett,2007,248(1):147-152.
[4]王奇,芦柏震.齐墩果酸的研究进展[J].中国药房,2008,19(9):711-712.
[5]Lin G,Nnane IP,Cheng T Y.The effects of pretreatment with glycyrrhizin and glycyrrhetinic acid on the retrorsineinduced hepatotoxicity in rats[J].Toxicon,1999,37 (9):1259-1270.
[6]Cinatl J,Morgenstern B,Bauer G,et al.Glycyrrhizin,an active component of liquorice roots,and replication of SARS-associated coronavirus[J].The Lancet,2003,361 (9374):2045-2046.
[7]Lee C S,Kim Y J,Lee M S,et al.18β-Glycyrrhetinic acid induces apoptotic cell death in SiHa cells and exhibits a synergistic effect against antibiotic anti-cancer drug toxicity[J].Life Sci,2008,83(13/14):481-489.
[8]Kalaiarasi P,Kaviarasan K,Pugalendi K V.Hypolipidemic activity of 18β-glycyrrhetinic acid on streptozotocininduced diabetic rats[J].Eur J Pharmacol,2009,612 (1/3):93-97.
[9]Kalaiarasi P,Pugalendi K V.Antihyperglycemic effect of 18β-glycyrrhetinic acid,aglycone of glycyrrhizin,on streptozotocin-diabetic rats[J].Eur J Pharmacol,2009,606 (1/3):269-273.
[10]汤旭蓁,关腾,钱贻崧,等.糖原磷酸化酶抑制剂山楂酸对小鼠血糖和肝糖原的影响[J].中国天然药物,2008,6(1):53-56.
[11]Reyes-Zurita F J,Rufino-Palomares E E,Lupianez JA,et al.Maslinic acid,a natural triterpene fromOlea europaeaL.,induces apoptosis in HT29 human colon-cancer cells via the mitochondrial apoptotic pathway[J].Cancer Lett,2009,273(1):44-54.
[12]Yan S L,Huang C Y,Wu S T,et al.Oleanolic acid and ursolic acid induce apoptos is in four human liver cancer cell lines[J].Toxicol In Vitro,2010,24(3):842-848.
[13]Fontanay S,Grare M,Mayer J,et al.Ursolic,oleanolic and betulinic acids:antibacterial spectra and selectivity indexes[J].JEthnopharmacol,2008,120(2):272-276.
[14]Tu H Y,Huang A M,Wei B L,et al.Ursolic acid derivatives induce cell cycle arrest and apoptosis in NTUB1 cells associated with reactive oxygen species[J].Bioorg Med Chem,2009,17(20):7265-7274.
[15]Liu W K,Ho JC K,Cheung FW K,et al.Apoptotic activity of betulinic acid derivatives onmurinemelanoma B16 cell line[J].Eur JPharmacol,2004,498(1/3):71-78.
[16]Gerrish D,Kim I C,Kumar D V,et al.Triterpene based compounds with potent anti-maturation activity against HIV-1[J].Bioorg Med Chem Lett,2008,18(24): 6377-6380.
[17]徐晓娣,孙辉辉,荣蓉,等.复方软胶囊中齐墩果酸在大鼠体内的药动学及相对生物利用度[J].中国药剂学杂志,2008,6(6):392-399.
[18]翟光喜,娄红祥,邹立家,等.药物磷脂复合物的研究进展[J].中国药物化学杂志,2001,36(12):800-803.
[19]蒋朝晖,宛蕾,陈秀芬.齐墩果酸二元酸单酯二钠的合成[J].中国药物化学杂志,1997,7(4):252-255.
[20]王文宇,陈济民.齐墩果酸磷酸酯二钠盐的药物动力学与生物利用度研究[J].沈阳药科大学学报,2001,18(6):402-405.
[21]Ma CM,Wu XH,Masao H,et al.HCV protease inhibitory,cytotoxic and apoptosis-inducing effects of oleanolic acid derivatives[J].J Pharm Pharmaceut Sci,2009,12 (2):243-248.
[22]艾宏儒,孟艳秋,赵娟.熊果酸衍生物的合成[J].当代化工,2008,37(3):238-240.
[23]申利红,赖宜生,张奕华,等.硝酸酯类甘草次酸衍生物的合成及抗肿瘤活性[J].中国药科大学学报,2008,39(2):103-107.
[24]Li JF,Zhao Y,CaiM M,etal.Synthesis and evaluation of a novel series of heterocyclic oleanolic acid derivatives with anti-osteoclast formation activity[J].Eur J Med Chem,2009,44(7):2796-2806.
[25]孙华,胡春,方唯硕.3-氧代齐墩果酸氨基酸偶联物的合成、水溶性的测定及抗肿瘤活性研究[J].中国药物化学杂志,2008,18(1):11-15.
[26]Jeong H J,ChaiH B,Park SY,etal.Preparation of amino acid conjugates of betulinic acid with activity against human melanoma[J].Bioorg Med Chem Lett,1999,9(8): 1201-1204.
[27]Huang D,Ding Y,Li Y,et al.Anti-tumor activity of a 3-oxo derivatives of oleanolic acid[J].Cancer Lett,2006,233(2):289-296.
[28]Meng Y Q,Liu D,Cai L L,et al.The synthesis of ursolic acid derivatives with cytotoxic activity and the investigation of their preliminary mechanism of action[J].Bioorg Med Chem,2009,17(2):848-854.
[29]Qian K D,Yu D L,Chen C H,et al.Anti-AIDS agents 78.Design,synthesis,metabolic stability assessment,and antiviral evaluation of novel betulinic acid derivatives as potent anti-human immunodeficiency virus(HIV)agents[J].JMed Chem,2009,52(10):3248-3258.
[30]Drag-Zalesinska M,Kulbacka J,Saczko J,et al.Esters of betulin and betulinic acid with amino acids have improved water solubility and are selectively cytotoxic toward cancer cells[J].Bioorg Med ChemLett,2009,19 (16):4814-4817.
[31]Parra A,Rivas F,Lopez P E,et al.Solution-and solidphase synthesis and anti-HIV activity ofmaslinic acid derivatives containing amino acids and peptides[J].Bioorg Med Chem,2009,17(3):1139-1145.
[32]臧静,李英霞,李春霞,等.四种齐墩果酸皂苷的合成[J].中国海洋大学学报,2005,35(4):635-640.
[33]Wang P,Wang J,Guo T T,et al.Synthesis and cytotoxic activity of theN-acetylglucosamine-bearing triterpenoid saponins[J].Carbohydr Res,2010,345(5):607-620.
[34]Huang Z J,Zhang Y H,Zhao L,et al.Synthesis and antihuman hepatocellular carcinoma activity of new nitric oxide-releasing glycosyl derivatives of oleanolic acid[J].Org Biomol Chem,2010,8(3):632-639.
[35]Gauthier C,Legault J,Rondeau S,et al.Synthesis of betulinic acid acyl glucuronide for application in anticancer prodrugmonotherapy[J].Tetrahedron Lett,2009,50(9): 988-991.
[36]Cheng K G,Liu J,Liu XF,et al.Synthesis of glucoconjugates of oleanolic acid as inhibitors of glycogen phosphorylase[J].Carbohydr Res,2009,344(7):841-850.
[37]张丽娟,吴常伟,钟家亮.L-精氨酸齐墩果酸复合物的制备与鉴定[J].中国药师,2009,12(10):1386-1388.
[38]许卉,刘生生,姜永涛,等.精氨酸甘草次酸的制备及核磁共振谱分析[J].分析化学,2006,34(5): 687-690.
[39]马金玲.齐墩果酸磷脂复合物及其制备方法:中国,1709283A[P].2005-12-21.
Advances in the Research on Preperation of Water-soluble Derivatives of Pentacyclic Triterpenoid Acid
SHANG Juan,CHEN Li
(DepartmentofPhytochemistry,ChinaPharmaceuticalUniversity,Nanjing210009,China)
Recent advances in the studies on derivatives of five kinds ofwater-soluble pentacyclic triter penoid acids has been reviewed.Pentacyclic triterpenoid acid possesses various biological activites.However,it is limited in the clinical application due to its poor solubility and low bioavailability.Therefore,it has become an interesting subject for the researchers to use pentacyclic triterpenoid acids as lead compounds to synthesize the water-soluble derivatives and to search for the candidate compounds having bioactivities and clinical value.
pentacyclic triterpenoid acid;water-soluble derivatives;biological activity
R 918
A
1001-5094(2011)05-0203-09
10.3969/j.issn.1001-5094.2011.05.002
[接受日期]2011-03-07
[项目资助]2009年中国药科大学中央高校基本科研业务费专项(No.JKY2009028)
*通讯作者:陈莉,副教授;
研究方向:天然药物化学;
Tel:025-83271447;E-mail:chenliduo12@gmail.com
(责任编辑:邢爱敏)
猜你喜欢
中草药(2022年16期)2022-08-16
作物学报(2022年7期)2022-05-12
中国食用菌(2022年4期)2022-05-11
皮肤病与性病(2021年3期)2021-07-30
药学进展(2021年3期)2021-05-11
药学进展(2021年3期)2021-05-11
健康博览(2021年4期)2021-04-23
西藏农业科技(2021年4期)2021-04-18
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
山西医科大学学报(2020年1期)2020-03-02
