
Theoretical Studies on Reaction and Kinetics of DNBF from TNAB
2011-01-28 03:03XIONGXianfengLUOYangLILi
火炸药学报 2011年5期
XIONG Xian-feng,LUO Yang,LI Li
(Xi′an Modern Chemistry Research Institute,Xi′an 710065,China)
Introduction
DNBF(4,6-dinitrobenzofuroxan)is the representative compound of energetic material which contain benzofuroxan ring.It is powerful in explosion and an important intermediate to synthesize other energetic materials.
There are several methods to prepare DNBF in the reported literature.Among those preparation methods,the method that DNBF is synthesized from 1-azido-2,4,6-trinitrobenze (TNAB)is friendly to environment for little waste water and waste acid[1-2].People pay much attention to this method from the techniques,reaction mechanism and reaction kinetics.The mechanism of decomposition ofσ-nitroazidobenzene with PM3-MO method was studied by Li Jin-shan et al[3]in 1998.In this paper,they studied the“furoxan mechanism”,the “breakage mechanism”of N—N2and the“cleavage mechanism”of C—NO2for the decomposition ofσ-nitroazidobenzene.In 1999,Li Jin-shan et al[4]reported the theoretical research of the reaction mechanism,thermodynamics and dynamics with PM3-MO calculation method.In 2000,a re-search on the stability and isomerization of benzofuroxan by B3LYP/6-31G(d)method and ab initio method was done by Li Jin-shan et al[5].In 2004,Zhou Hong-ping et al[6]studied the structure of aminonitrobenzodifuroxan with B3LYP/6-31G**method.
From the viewpoint of quantum chemistry,we studied the synthesis mechanism and reaction kinetic in this paper.The calculation results can provide valuable theoretic evidence for the synthesis and application for the DNBF.
1 Methodology
1.1 Electronic structure calculations
The geometries of all stationary points(reactants,products,and the transition state)are optimized by using B3LYP with the cc-pVTZ[7]basis sets.Frequency analyses of reactants,products and transition state were performed by B3LYP with the cc-pVTZ basis sets.All stationary points(reactants and products)had positive computed frequencies,while the transition state had only one imaginary frequency.The minimum energy path(MEP)was obtained using the intrinsic reaction coordinate(IRC)[8]theory at B3LYP/cc-pVTZ level by the step length 0.02(amu)1/2 bohr.All electronic structure calculations were carried out with the Gaussian 03program[9].
1.2 The rate constant calculations
The rate constants were computed using conventional transition-state theory (TST)[10],the TST rate constant calculations with the Eckart tunneling correction(TST/Eckart)[11]and RRKM(T)rate constant calculations with the Eckart tunneling correction(RRKM(T)/Eckart)[12].All the calculations were carried out with the online Vklab program package[13]over a wide temperature region from 200to 2500K.
2 Results and discussions
2.1 The optimized geometries of stationary points
The optimized geometric parameters of the reactant,products and transition state of the reaction were obtained by using B3LYP method with the ccpVTZ basis set.Table 1 lists the optimized geometric parameters of the equilibrium and transition state.

Table 1 The bond length of reactants,products,and transition state by using B3LYP/cc-pVTZ method
The optimized structures of the reactant,products,and transition states are shown in Fig.1 .

Fig.1 Optimized structures of the reactant(TNAB),transition states,products(DNBF and N2)and corresponding atomic number
The bond length,dihedral angle and bond an-gle of the optimized structure of the TNAB,TS and DNBF was shown in Table 1 and Table 2,respectively.
As shown in the Table 1,compared the bond length of the reactant,transition state and product,along the reaction path,new bond of O(9)—N(10)is gradually formed and a new furoxan ring is built,the bond lengh of C(1)—C(2)and N(7)—O(9)is shifted form 1.419Åand 1.224Åto 1.462Åand 1.426Å.The bond length of C(1)—N(7)and C(2)—N(10)decrease to 1.348 Åand 1.315 Å,respectively.The bond between N(10)—N(11)become weaker until to break completely,so the bond N(11)—N(12)can leave from the DNBF and form N2molecule.

Table 2 The dihedral angle and bond angle of reactants,products and transition state using B3LYP/cc-pVTZ method
From the dihedral angle in the Table 2,it is indicated that,the-NO2(O(8)—N(7)—O(9)),-N3(N(10)—N(11)—N(12))group in the reactant,transition state are deviated from the plane formed by benzene ring.In the transition state,the C(1)—C(2)—N(7)—O(9)—N(10)are formed furazan ring and in a plane with the benzene ring,the atom O(8)is in a plane with the furazan ring.Another two-NO2groups formed by O(19)—N(17)—O(18),O(15)—N(13)—O(14)deviate from the benzene ring plane along the reaction course.
In our synthetical experiment,the DNBF single crystal have been obtained and characterized by X-ray diffraction analysis.Compared the calculated data with the crystal data in the Table 1,the results indicated that the calculated data are consistent with the crystal data[1].
2.2 Frequencies
The infrared spectra of TNAB and DNBF which calculated by B3LYP/cc-pVTZ method are shown in Fig.2 and Fig.3 .
As shown in Fig.2,the most intense vibrations in the IR spectrum are N—N—N asymmetric vibrations at 2 250 ~2 290.The adsorption of TNAB at 738and 1 493cm-1may be corresponding to the vibration of benzene ring.The vibration at 947cm-1is raised by the stretch vibration of C—N bond,the adsorption at 1 094cm-1may be corresponding to the bend vibration of the C—H bond which is connected to the benzene ring,The adsorption at 1 365,1 370and 1 387cm-1are raised by the symmetric stretch vibration of C—H bond,the adsorption at 1 582,1 601and 1 616cm-1may be corresponded to the asymmetric stretch vibration of N—O bond.

Fig.2 The IR spectra plot of TNAB calculated at B3LYP/cc-pVTZ level of theory
As shown in Fig.3,the absorption at 709cm-1may be corresponding to the bend vibration of C—H bond that is connected to the benzene ring,the absorption at 758cm-1indicate the existence of C—N—O bond,the absorption at 829cm-1may corresponding to the stretch vibration of C—N bond,the adsorption at 1 369and 1 531cm-1are raised by the stretch vibration and asymmetric stretch vibration of N—O bond,respectively.The adsorption at 1 484and 1 623cm-1may be corresponding to the stretch vibration of furoxan group.
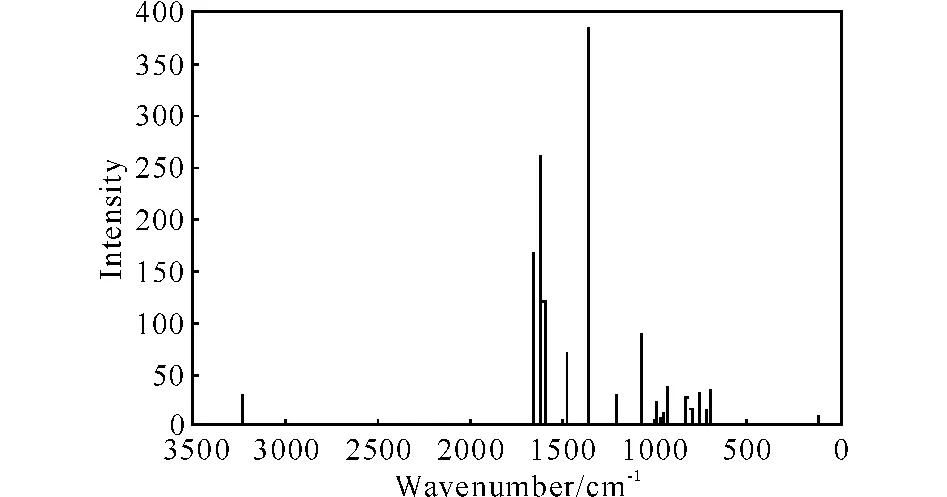
Fig.3 The IR spectra plot of DNBF calculated at B3LYP/cc-pVTZ level of theory
Compared the calculated IR spectra with the experimental spectra[1]obtained by FT-IR with KBr pellet(ν:1334,1540,NO2;ν:3079,Ar-H;ν:1623,1451,benzofuroxan ring),we can found that the two kinds of IR spectra are consistent well.
2.3 Minimum energy path
The minimum-energy paths of the reaction were obtained using the intrinsic reaction coordinate theory at the B3LYP/cc-pVTZ level and the potential energy profile was obtained.Fig.4 describe the vibrationally adiabatic ground-state potential curve(VaG)of the reaction as a function of s(amu)1/2using B3LYP method with cc-pVTZ basis set.It can be seen that for the(VaG)curve at this level is an ideal potential reaction surface.

Fig.4 The IRC result calculated by B3LYP/cc-pVTZ
The classical reaction energy barrier and reaction enthalpies for the TNAB→DNBF + N2are 120.80and -14.09kJ/mol,respectively.With the zero-point energy correction,the reaction barrier and reaction enthalpies are 113.36and-21.78 kJ/mol,respectively.
2.4 Reaction kinetics
The reaction rate constants was calculated using the TST,TST/Eckart and RRKM(T)/Eckart method based on the interpolated MEP at the B3LYP/cc-pVTZ levels of the theory.The quantum tunneling effects are included in the rate constants calculations with the Eckart tunneling model.
The rate constants of the reaction of TNAB→DNBF+ N2are shown in Fig.5 .

Fig.5 Arrhenius plots of the reaction rate constants calculated at the TST,TST/Eckart and RRKM(T)/Eckart levels of theory
From the Fig.5,the reaction rate calculated by TST,TST/Eckart and RRKM(T)/Eckart is equal well during the high temperature range(T>600K),which indicate that the tunnel effect and RRKM theory have no influence in the high temperature range.In the low temperature range(200K <T<600K),the consistency between TST/Eckart and RRKM(T)/Eckart method is very well and the difference with TST method is become large along the decline of temperature.It is indicated that the influence of tunnel and the RRKM theory to the reaction is obvious in the low temperature range.
For this reaction of TNAB→DNBF + N2,the Arrhenius rate equation can be described by the equation(1):

For the reaction of TNAB→DNBF+N2,according to the calculated results,the fitted threeparameter Arrhenius rate equation can be described as follows,respectively.

3 Summary
(1)A direct dynamics study for preparing DNBF from TNAB was carried out by B3LYP/ccpVTZ level of the theory.The geometries parameter and harmonic vibrational frequencies of all stationary points were calculated with the same level of theory.The minimum energy path was calculated by the IRC theory.The rate constants were evacuated using TST,TST/Eckart and RRKM (T)/Eckart in the temperature range of 200-2 500K.
(2)According to the calculated results,we can conclude the tunneling effect is very important in the reaction.It was found that the rate constant from the TST/Eckart and RRKM (T)/Eckart method is very consistent.The tunneling effect and RRKM theory will have some effects on the reaction in practice.Thus this study provides some valuable information for the reaction route of preparing DNBF by direct dynamics study.
Reference:
[1]苗艳玲,张同来,乔小晶,等.4,6-二硝基苯并氧化呋咱的制备、晶体结构及热分解机理[J].有机化学,2004,24(2):205-209.
MIAO Yan-ling,ZHANG Tong-lai,QIAO Xiao-jing,et al.Preparation,crystal structure and thermal decomposition mechanisms of 4,6-dinitrobenzofuroxan[J].Chinese Journal of Organic Chemistry,2004,24(2):205-209.
[2]Reddy G O,Muralib K M,Chatterjee A K.Thermal study on picryl azide(2-azido-1,3,5-trinitrobenzene)decomposition using simultaneous thermogravimetry and differential scanning calorimetry[J].Propellants,Explosives,Pyrotechnics,1983,8(1):29-33.
[3]李金山,肖鹤鸣,陈兆旭,等.邻硝基叠氮苯分解机理的PM3-MO 研究[J].分子科学学报,1998,14(3)193-197.
LI Jin-shan,XIAO He-ming,CHEN Zhao-xu,et al.A study on the mechanism of decomposition of o-nitroazidobenzene with PM3-MO method[J].Journal of Molecular Science,1998,14(3):193-197.
[4]李金山,肖鹤鸣,贡雪东,等.2-叠氮-1,3,5-三硝基苯热解反应的机理及其热力学和动力学理论研究[J].爆炸与冲击,1999,19(1):39-42.
LI Jin-shan,XIAO He-ming,GONG Xue-dong,et al.Theoretical study on the mechanism,thermodynamics and kinetics of 2-azido-1,3,5-trinitrobenzene thermolysis[J].Explosion and Shock Waves,1999,19(1):39-42.
[5]李金山,肖鹤鸣,董海山.苯并氧化呋咱稳定性和异构化的DFT 和ab initio研究[J].化学物理学报,2000,13(1):55-60.
LI Jin-shan,XIAO He-ming,DONG Hai-shang.DFT and Ab initio studies on stablility and isomerization of benzofuroxan[J].Chinese Journal of Chemical Physical,2000,13(1):55-60.
[6]周红萍,董海山,郝莹,等.氨基硝基苯并二氧化呋咱结构的密度泛函理论研究[J].爆炸与冲击,2004,24(5):396-399.
ZHOU Hong-ping,DONG Hai-shan,HAO Ying,et al.Density functional theory study on the structure of aminonitrobenzodifuroxan[J].Explosion and Shock Waves,2004,24(5):396-399.
[7]Dunning J,Thom H.Gaussian basis sets for use in correlated molecular calculations[J].Journal of Chemical Physics,1989,90(2):1007-1022.
[8]Gracia L,Andrés J,Safont V S,et al.DFT study of the reaction between VO2and C2H6[J].Organometallics,2004,23(4):730-739.
[9]Frisch M J,Trucks G W,Schlegel H B.Gaussian 03.Revision A.01.[M].Pittsburgh,PA:Gaussian Inc,2003.
[10]González M,Miquel I,Sayós R.VTST kinetics study of the N+O2→NO+O reactions based on CASSCF and CASPT2:Ab initio calculations including excited potential energy surfaces[J].Chemical Physics Letters,2001,335(3-4):339-347.
[11]Kerkeni B,Clary D C.Quantum scattering study of abstraction reactions of H atoms from CH3NH2[J].Chemical Physics Letters,2007,438(1/3):1-7.
[12]Tarrazo-Antelo T,Martínez-Núňez E,Vázquez S A.Ab initio and RRKM study of the elimination of HF and HCL from chlorofluoroethylene[J].Chemical Physics Letters,2007,435(4/6):176-181.
[13]Zhang Shao-wen,Truong T N.VKLab version 1.0[CP/CD].Utah:University of Utah,2001.
猜你喜欢
云南化工(2022年9期)2022-10-12
含能材料(2021年5期)2021-06-03
能源化工(2021年6期)2021-02-26
黄河之声(2021年19期)2021-02-24
书香两岸(2020年3期)2020-06-29
商品与质量(2019年32期)2019-11-29
火工品(2018年1期)2018-05-03
现代农业科技(2018年23期)2018-02-18
青年文学家(2016年34期)2017-03-31
安徽农业科学(2015年9期)2015-01-12
